Vorlage:Reactionbox The Pinnick oxidation is an organic reaction by which aldehydes can be oxidized into their corresponding carboxylic acids using sodium chlorite (NaClO2) under mild acidic conditions. It was originally developed by Lindgren and Nilsson.[1] The typical reaction conditions used today were developed by G. A. Kraus.[2][3] H.W. Pinnick later demonstrated that these conditions could be applied to oxidize α,β-unsaturated aldehydes.[4] There exist many different reactions to oxidize aldehydes, but only a few are amenable to a broad range of functional groups. The Pinnick oxidation has proven to be both tolerant of sensitive functionalities and capable of reacting with sterically hindered groups. This reaction is especially useful for oxidizing α,β-unsaturated aldehydes, and another one of its advantages is its relatively low cost.[4][5]

PinnickOxidationReaction

Mechanism
BearbeitenThe proposed reaction mechanism involves chlorous acid as the active oxidant, which is formed under acidic conditions from chlorite.
- ClO2− + H2PO4− Vorlage:Eqm HClO2 + HPO42−
First, the chlorous acid adds to the aldehyde. Then resulting structure undergoes a pericyclic fragmentation in which the aldehyde hydrogen is transferred to an oxygen on the chlorine, with the chlorine group released as hypochlorous acid (HOCl).[6]

Side reactions and scavengers
BearbeitenThe HOCl byproduct, itself a reactive oxidizing agent, can be a problem in several ways.[6] It can destroy the NaClO2 reactant:
- HOCl + 2ClO2− → 2ClO2 + Cl− + OH−
making it unavailable for the desired reaction. It can also cause other undesired side reactions with the organic materials. For example, HOCl can react with double bonds in the organic reactant or product via a halohydrin formation reaction.
To prevent interference from HOCl, a scavenger is usually added to the reaction to consume the HOCl as it is formed. For example, one can take advantage of the propensity of HOCl to undergo this addition reaction by adding a sacrificial alkene-containing chemical to the reaction mixture. This alternate substrate reacts with the HOCl, preventing the HOCl from undergoing reactions that interfere with the Pinnick reaction itself. 2-Methyl-2-butene is often used in this context:

Resorcinol and sulfamic acid are also common scavenger reagents.[6][7]
Hydrogen peroxide (H2O2) can be used as HOCl scavenger whose byproducts do not interfere in the Pinnick oxidation reaction:
- HOCl + H2O2 → HCl + O2 + H2O
In a weakly acidic condition, fairly concentrated (35%) H2O2 solution undergoes a rapid oxidative reaction with no competitive reduction reaction of HClO2 to form HOCl.
- HClO2 + H2O2 → HOCl + O2 + H2O
Chlorine dioxide reacts rapidly with H2O2 to form chlorous acid.
- 2ClO2 + H2O2 → 2HClO2 + O2
Also the formation of oxygen gives good indication of the progress of the reaction. However, problems sometimes arise due to the formation of singlet oxygen in this reaction, which may oxidize organic materials (i.e. the Schenck ene reaction). DMSO has been used instead of H2O2 to oxidize reactions that do not produce great yields using only H2O2. Mostly electron rich aldehydes fall under this category.[7] (See Limitation below)
Also, solid-supported reagents such as phosphate-buffered silica gel supported by potassium permanganate and polymer-supported chlorite have been prepared and used to convert aldehydes to carboxylic acid without having to do conventional work-up procedures. The reaction involves the product to be trapped on silica gel as their potassium salts. Therefore, this procedure facilitates easy removal of neutral impurities by washing with organic solvents.[8]
Scope and limitations
BearbeitenThe reaction is highly suited for substrates with many group functionalities. β-aryl-substituted α,β-unsaturated aldehydes works well with the reaction conditions. Triple bonds directly linked to aldehyde groups or in conjugation with other double bonds can also be subjected to the reaction.[7][9] Hydroxides, epoxides, benzyl ethers, halides including iodides and even stannanes are quite stable in the reaction.[7][9][10][11] The examples of the reactions shown below also show that the stereocenters of the α carbons remain intact while double bonds, especially trisubsituted double bonds do not undergo E/Z–isomerization in the reaction.
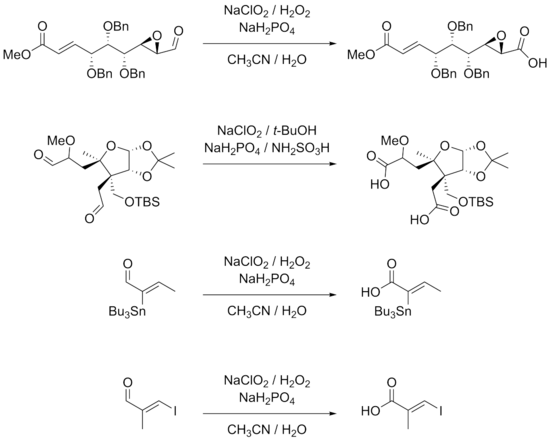
Scope

Lower yields are obtained for reactions involving aliphatic α,β-unsaturated and more hydrophilic aldehydes. Double bonds and electron-rich aldehyde substrates can lead to chlorination as an alternate reaction. The use of DMSO in these cases gives better yield. Unprotected aromatic amines and pyrroles are not well suited for the reactions either. In particular, chiral α-aminoaldehydes do not react well due to epimerization and because amino groups can be easily transformed to their corresponding N-oxides. Standard protective group approaches, such as the use of t-BOC, are a viable solution to these problems.[12]
Thioethers are also highly susceptible to oxidation. For example, Pinnick oxidation of thioanisaldehyde gives a high yield of carboxylic acid products, but with concomitant conversion of the thioether to the sulfoxide or sulfone.[7]

See also
BearbeitenEinzelnachweise
Bearbeiten- ↑ Lindgren, Bengt O., Torsten Nilsson, Steinar Husebye, ØYvind Mikalsen, Kurt Leander, Carl-Gunnar Swahn: Preparation of Carboxylic Acids from Aldehydes (Including Hydroxylated Benzaldehydes) by Oxidation with Chlorite. In: Acta Chem. Scand. 27. Jahrgang, 1973, S. 888–890, doi:10.3891/acta.chem.scand.27-0888.
- ↑ George A. Kraus, Bruce Roth: Synthetic studies toward verrucarol. 2. Synthesis of the AB ring system. In: J. Org. Chem. 45. Jahrgang, Nr. 24, 1980, S. 4825–4830, doi:10.1021/jo01312a004 (iastate.edu).
- ↑ George A. Kraus, Michael J. Taschner: Model studies for the synthesis of quassinoids. 1. Construction of the BCE ring system. In: J. Org. Chem. 45. Jahrgang, Nr. 6, 1980, S. 1175–1176, doi:10.1021/jo01294a058 (iastate.edu).
- ↑ a b Bal, B. S., Childers, W.E., Pinnick, H.W.: Oxidation of α,β-Unsaturated Aldehydes. In: Tetrahedron. 37. Jahrgang, Nr. 11, 1981, S. 2091–2096, doi:10.1016/S0040-4020(01)97963-3.
- ↑ B. J. Mundy, Michael G. Ellerd, Frank G. Favaloro: Name Reactions and Reagents in Organic Synthesis. John Wiley & Sons, 2005, ISBN 978-0-471-22854-7, S. 518.
- ↑ a b c László Kürti, Barbara Czakó: Strategic applications of named reactions in organic synthesis: background and detailed mechanisms. Elsevier, 2005, ISBN 978-0-12-429785-2, S. 354–356.
- ↑ a b c d e Dalcanale, E, Montanari, F: Selective Oxidation of Aldehydes to Carboxylic Acids with Sodium Chlorite-Hydrogen Peroxide. In: J. Org. Chem. 51. Jahrgang, Nr. 4, 1986, S. 567–569, doi:10.1021/jo00354a037.
- ↑ Takemoto, T., Yasuda, K., Ley, S.V.: Solid-Supported Reagents for the Oxidation of Aldehydes to Carboxylic Acids. In: Synlett. 2001. Jahrgang, Nr. 10, 2001, S. 1555–1556, doi:10.1055/s-2001-17448.
- ↑ a b Raach, A., Reiser, O.: Sodium Chlorite-Hydrogen Peroxide, a Mild and Selective Reagent for the Oxidation of Aldehydes to Carboxylic Acids. In: J. Prakt. Chem. 342. Jahrgang, Nr. 6, 2000, S. 605–608, doi:10.1002/1521-3897(200006)342:6<605::aid-prac605>3.0.co;2-i.
- ↑ Ishihara, J., Hagihara, K., Chiba, H., Ito, K., Yanagisawa, Y., Totani, K, Tadano, K.: Synthetic studies of viridenomycin. Construction of the cyclopentene carboxylic acid part. In: Tetrahedron Lett. 41. Jahrgang, Nr. 11, 2000, S. 1771–1774, doi:10.1016/S0040-4039(00)00013-7.
- ↑ Kuramochi, K., Nagata, S., Itaya, H., Takao, H., Kobayashi, S.: Convergent Total Synthesis of epolactaene: application of bridgehead oxiranyl anion strategy. In: Tetrahedron Lett. 40. Jahrgang, Nr. 41, 1999, S. 7371–7374, doi:10.1016/S0040-4039(99)01512-9.
- ↑ Dehoux, C., Fontaine, E., Escudier, J., Baltas, M., Gorrichon, L.: Total Synthesis of Thymidine 2-Deoxypolyoxine C Analogue. In: J. Org. Chem. 63. Jahrgang, Nr. 8, 1998, S. 2601–2608, doi:10.1021/jo972116s, PMID 11672125.
[[Category:Name reactions]] [[Category:Organic oxidation reactions]]