Meningeom
| Klassifikation nach ICD-10 | |
|---|---|
| D32 | Gutartige Neubildung der Meningen |
| D32.0 | Hirnhäute |
| D32.1 | Rückenmarkhäute |
| D32.9 | Meningen, nicht näher bezeichnet |
| C70 | Bösartige Neubildung der Meningen |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9530/0 | Meningeom o.n.A. Lymphoplasmozytenreiches Meningeom Metaplastisches Meningeom Mikrozystisches Meningeom Sekretorisches Meningeom |
| 9530/3 | Malignes Meningeom Anaplastisches Meningeom Leptomeningeales Sarkom Meningeales Sarkom Meningotheliales Sarkom |
| ICD-O-3, zweite Revision (2019) | |
Ein Meningeom (synonym: Meningiom, griechisch-lateinisch: Meningeoma, englisch: meningioma) ist ein meistens benigner („gutartiger“) intrakranieller Tumor. Er entsteht durch die Entartung von Zellen der Arachnoidea (einer Hirnhautschicht). Charakteristisch ist sein langsames und verdrängendes Wachstum. Maligne („bösartige“) Entartungen sind selten. Der Oberbegriff ist Dignität. 20 bis 25 % aller primären Tumoren des Zentralnervensystems sind Meningeome.
Vorkommen
BearbeitenDas Haupterkrankungsalter liegt im 5. Lebensjahrzehnt, wobei Frauen im Verhältnis 3:2 häufiger betroffen sind. Ein multiples Auftreten von Meningeomen ist charakteristisch für die Neurofibromatose Typ 2. In bis zu 19 % der Fälle treten mehrere Läsionen gleichzeitig auf (Multiple Meningiome, Meningiomatosis).[1][2]
Ursachen
BearbeitenDie meisten Meningeome entstehen sporadisch. Am häufigsten finden sich genetische Veränderungen bestimmter Tumorsupressorproteine, von denen ein Verlust von NF2 in bis zu 60 % der Fälle beobachtet wird.[3] Bestimmte Genveränderungen treten nur in bestimmten morphologischen Varianten auf, so finden sich KLF4 (K409Q)-Punktmutationen nur bei sekretorischen Meningeomen,[4] während AKT1 (E17K)-Mutationen mit meningeothelialen und transitionalen Varianten assoziiert sind.[5] Meningeome können auch als Spätfolgen einer vorausgegangenen Strahlentherapie auftreten. Zahnärztliche Röntgendiagnostik in der Vorgeschichte scheint abhängig vom Alter zum Zeitpunkt der Aufnahme und der verwendeten Röntgentechnik das Risiko, an einem Meningeom zu erkranken, erhöhen zu können.[6]
Einteilung
BearbeitenNach der WHO-Klassifikation der Tumoren des zentralen Nervensystems sind derzeit folgende 3 Tumorgrade bekannt, die sich in Rezidivhäufigkeit und Prognose unterscheiden:[7]
- Grad I (gutartig), 80–90 %, pleomorph, operativ meist komplett entfernbar, gute Prognose,
- Grad II (atypisch), 5–15 %, schnelleres Wachstum, häufiger Rezidive
- Grad III (anaplastisch), 1–3 %, bösartig, infiltratives Wachstum. Zu dieser Gruppe gehören anaplastisches Meningeom, leptomeningeales Sarkom, meningeales Sarkom und meningotheliales Sarkom
Pathologie
Bearbeiten

Meistens befinden sich Meningeome an der Falx cerebri, am Keilbeinflügel, an der Olfaktoriusrinne und setzen sich in der Regel gut vom angrenzenden Hirngewebe ab. Die Schnittfläche sieht grau und körnig aus. In manchen Formen ist im Mikroskop eine Zwiebelschalenformation der Tumorzellen zu beobachten. Verkalken diese, werden sie Psammomkörper genannt.
Makroskopie: abgekapselte, rundliche, grau-weiße Tumoren von prall-elastischer und derber Konsistenz. Sie heften fest der Dura an und komprimieren das angrenzende Hirngewebe. Häufig kommt eine Dura- und/oder Knocheninfiltration mit Hyperostose des anliegenden Knochens vor. Die Meningeome sind hypervaskularisiert, es können sich Furchen in den Knochen bilden (siehe Sulci arteriosi durch Arteria meningea media).
Histologie: Bestimmte morphologische Varianten werden direkt einem bestimmten WHO-Grad zugeordnet. So werden meningeotheliomatöse, fibromatöse, sekretorische, mikrozytische, transitionale, psammomatöse und angiomatöse Meningeome zu den WHO-Grad-I-Tumoren gezählt. Das chordoide und das klarzellige Meningeom entsprechen einem WHO Grad II, während papilläre und rhabdoide Meningeome zu den WHO-Grad-III-Meningeomen gerechnet werden.
Symptome
BearbeitenZu den geschilderten Beschwerden gehören Kopfschmerzen und neurologische Ausfälle. In der Schwangerschaft kann das Wachstum der Meningeome beschleunigt verlaufen, eine mögliche Erklärung sind die in den Tumorzellen regelmäßig vorhandenen Progesteron-Rezeptoren. Zufällig entdeckte Meningeome können auch gar keine Symptome hervorrufen und müssen, wenn sie nicht rasch größer werden, nicht unbedingt operiert werden.
Diagnostik
BearbeitenDas bildgebende Verfahren der ersten Wahl ist beim Meningeom heute die Kernspintomografie, da dieses Verfahren den größten Weichteilkontrast besitzt und in typischen Fällen die sichere Diagnose eines Meningeoms ermöglicht. In T2-gewichteten Aufnahmen stellen sich verkalkte Meningeome im Gegensatz zu vielen anderen Tumoren als schwarze Masse (hypointens) dar, die dunkler als das umgebende Hirngewebe ist. Unverkalkte Meningeome können zur Umgebung isointens sein. Von anderen Tumoren unterscheiden sich Meningeome durch ihre Lage auf der Dura mater mit charakteristischen Ausläufern in die Dura (dural tails) und durch eine sehr intensive Kontrastmittelaufnahme. Die Computertomographie kann die Tumorverkalkungen sehr gut nachweisen. Konventionelles Röntgen und Angiographie haben heute nur noch eine untergeordnete Bedeutung.
Die meisten Meningeome wachsen sphärisch oder globulär unter Beibehaltung ihrer soliden Masse. In manchen Fällen können sie Hirnhäute, Knochen oder auch venöse Blutleiter durchbrechen. Liegt eine Infiltration des Sinus sagittalis superior vor, werden die Meningeome in 6 verschiedene Subtypen nach Sindou eingeteilt.[8] Daneben beobachtet man bevorzugt im Os sphenoidale eine plaqueförmige Ausbreitung.
-
 Meningeom im Röntgenbild des Schädels
Meningeom im Röntgenbild des Schädels -
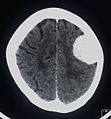 Kontrastmittel aufnehmendes Meningeom der linken Hemisphäre in der Computertomographie
Kontrastmittel aufnehmendes Meningeom der linken Hemisphäre in der Computertomographie -
 Großes Felsenbeinmeningeom in der Computertomographie
Großes Felsenbeinmeningeom in der Computertomographie -
 Typische randständige Lokalisation eines Meningeoms rechts (Kernspintomographie, T1-gewichtet mit Kontrastmittel)
Typische randständige Lokalisation eines Meningeoms rechts (Kernspintomographie, T1-gewichtet mit Kontrastmittel) -
 Falxmeningeom in der Kernspintomographie: T1 mit Kontrastmittel
Falxmeningeom in der Kernspintomographie: T1 mit Kontrastmittel -
 Meningeom im Spinalkanal Kernspintomographie. Links T1 sagittal mit Kontrastmittel, rechts T2 coronar
Meningeom im Spinalkanal Kernspintomographie. Links T1 sagittal mit Kontrastmittel, rechts T2 coronar






Therapie
BearbeitenDie neurochirurgische Entfernung des Tumors ist die Therapie der Wahl. Eine Option kann hier bei gefäßreichen Tumoren die präoperative Embolisation sein. Eventuell kann auch eine Strahlentherapie oder Radiochirurgie (Gamma-Knife oder Cyberknife) durchgeführt werden. Kann ein Meningeom nicht komplett operativ entfernt werden, so kann auch nach der Operation eine Bestrahlung der verbliebenen Tumorreste erfolgen, um die Wahrscheinlichkeit für ein erneutes Tumorwachstum zu reduzieren. Kleine Meningeome ohne Wachstumstendenz bei älteren Personen müssen nicht unbedingt entfernt werden.
Insbesondere bei größeren Meningeomen mit komplexer (nicht kugeliger) Form und Nahebeziehung zu strahlensensiblen Organen wie z. B. Sehnerv oder Hypophyse (Hirnanhangsdrüse, steuert die Hormondrüsen im Körper) empfiehlt sich die schonendere fraktionierte stereotaktische Bestrahlung.[9] Bei der stereotaktischen Präzisionsbestrahlung per Linearbeschleuniger erzeugt man Röntgenstrahlung. Durch einen softwaregesteuerten Mikro-Multi-Leaf-Kollimator (MMLC) wird die Bestrahlungsdosis an die Form des Tumors angepasst. Bei dem Multi-Leaf-Kollimator handelt es sich um bewegliche Metallblenden von je 3 mm Dicke, die individuell verstellbar sind und so gesundes Gewebe verdecken und schützen können.[10] Die Lagerung erfolgt hier nicht wie bei der Gamma-knife-Bestrahlung über einen Metallrahmen, der am Kopf angeschraubt wird, sondern über eine spezielle Maske (thermoplastische Kopfmaske), die individuell anmodelliert und vor jeder Behandlungssitzung aufgesetzt wird.[11] Diese Form der Hochpräzisionsbestrahlung wird in speziell ausgerüsteten Abteilungen für Strahlentherapie angeboten. Der Vorteil der Aufteilung der Bestrahlung auf mehrere Sitzungen liegt in der besseren DNA-Reparaturmöglichkeit der Zellen gesunder Gewebe in den Pausen zwischen zwei Bestrahlungssitzungen. Durch diese Erholung (genannt Fraktionierungseffekt) kann das Risiko für schwerwiegende Nebenwirkungen durch die Bestrahlung zusätzlich gesenkt werden.[12]
Neuere Ansätze in der Behandlung von Meningeomen beschäftigen sich mit dem zusätzlichen Einsatz von Blutgefäßwachstum bzw. Tumorwachstum hemmenden Medikamenten. Hierbei wird versucht insbesondere bei wiederkehrenden aggressiveren Meningeomen, die nicht mehr operiert oder erneut bestrahlt werden können, zu behandeln. Dieser Ansatz ist aber noch experimentell und kein Standardverfahren.[13]
Geschichte
BearbeitenIm 18. und 19. Jahrhundert wurden Meningeome als Fungus bezeichnet. Erstmals genauer beschrieben wurden diese Tumoren 1829 von Jean Cruveilhier. Den Begriff Meningeom prägte Harvey Cushing in seiner mit Louise Eisenhardt 1938[14] veröffentlichten Monografie Meningeomas. Rudolf Virchow nahm 1864 an, dass es sich um Sarkome handelt, weswegen man die Mengingeome zunächst für bösartig hielt. Im Jahr 1887 gelang W. W. Keen in den USA erfolgreich die operative Entfernung eines Meningeoms.[15]
Literatur
Bearbeiten- Joung H. Lee (Hrsg.): Meningiomas: Diagnosis, Treatment, and Outcome. Springer, London 2008, ISBN 978-1-84628-526-4.
- C. Mawrin, A. Perry: Pathological classification and molecular genetics of meningiomas. In: Journal of Neuro-Oncology. Band 99, Nummer 3, September 2010, S. 379–391. doi:10.1007/s11060-010-0342-2. PMID 20809251. (Review).
- I. Whittle, C. Smith, P. Navoo, D. Collie: Meningiomas. In: The Lancet. 363, 2004, S. 1535–1543, doi:10.1016/S0140-6736(04)16153-9.
- Anne G Osborn: Osborn’s Brain : Imaging, pathology and anatomy. Amirsys Publishing, 2013, ISBN 978-1-931884-21-1.
- M. Kufeld, B. Wowra, A. Muacevic, S. Zausinger, J. C. Tonn: Radiosurgery of spinal meningiomas and schwannomas. In: Technology in cancer research & treatment. Band 11, Nummer 1, Februar 2012, S. 27–34, doi:10.7785/tcrt.2012.500231, PMID 22181328, PMC 4527413 (freier Volltext).
Weblinks
Bearbeiten- Meningeome, Deutsche Hirntumorhilfe e. V.
- meningeom.at, Österreichische Selbsthilfegruppe Meningeom, Liste behandelnder Krankenhäuser und Ärzte.
Einzelnachweise
Bearbeiten- ↑ Andres Ramos-Fresnedo, Ricardo A. Domingo, Tito Vivas-Buitrago, Larry Lundy, Daniel M. Trifiletti: Multiple meningiomas: does quantity matter? a population-based survival analysis with underlined age and sex differences. In: Journal of Neuro-Oncology. Band 149, Nr. 3, September 2020, ISSN 1573-7373, S. 413–420, doi:10.1007/s11060-020-03620-7, PMID 32986140.
- ↑ J. O. Lusins, H. Nakagawa: Multiple meningiomas evaluated by computed tomography. In: Neurosurgery. Band 9, Nr. 2, August 1981, ISSN 0148-396X, S. 137–141, doi:10.1227/00006123-198108000-00004, PMID 7266810.
- ↑ L. R. De Vitis, A. Tedde u. a.: Screening for mutations in the neurofibromatosis type 2 (NF2) gene in sporadic meningiomas. In: Hum Genet. 97(5), Mai 1996, S. 632–637.
- ↑ D. E. Reuss, R. M. Piro u. a.: Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. In: Acta Neuropathol. 125(3), Mar 2013, S. 351–358.
- ↑ F. Sahm, J. Bissel u. a.: AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. In: Acta Neuropathol. 126(5), Nov 2013, S. 757–762.
- ↑ Elizabeth B. Claus, Lisa Calvocoressi u. a.: Dental x-rays and risk of meningioma. In: Cancer. Band 118, Nr. 18, 15. September 2012, S. 4530–4537, doi:10.1002/cncr.26625.
- ↑ NOA - Neuroonkologische Arbeitsgemeinschaft. Abgerufen am 28. Dezember 2020.
- ↑ Sindou Klassifikation für Meningeome - Ars Neurochirurgica
- ↑ A. E. Elia, H. A. Shih, J. S. Loeffler: Stereotactic radiation treatment for benign meningiomas. In: Neurosurgical Focus. 23(4), 2007, S. E5. PMID 17961042.
- ↑ Archivierte Kopie ( vom 14. Juli 2014 im Internet Archive)
- ↑ Julia Ahlswede: Aspekte der Positionierung und Dosisapplikation in der stereotaktisch geführten intra- und extrakranialen Strahlentherapie. 2005. (edoc.hu-berlin.de)
- ↑ STEREOTACTIC RADIOSURGERY AND RADIOTHERAPY OF MENINGIOMAS. Clinical White Paper. PDF-Version
- ↑ Emil Lou, Ashley L. Sumrall u. a.: Bevacizumab therapy for adults with recurrent/progressive meningioma: a retrospective series. In: J Neurooncol. 109(1), Aug 2012, S. 63–70. PMC 3404217 (freier Volltext); Quelle der Übersetzung
- ↑ Cushing, Eisenhardt: Meningeomas. Thomas, Springfield, Ill./ Baltimore 1938.
- ↑ Wolfgang Seeger, Carl Ludwig Geletneky: Chirurgie des Nervensystems. In: Franz Xaver Sailer, Friedrich Wilhelm Gierhake (Hrsg.): Chirurgie historisch gesehen. Anfang – Entwicklung – Differenzierung. Dustri-Verlag, Deisenhofen bei München 1973, ISBN 3-87185-021-7, S. 229–262, hier: S. 239.