Enzymkinetik
Die Enzymkinetik ist ein Teilgebiet der biophysikalischen Chemie. Sie beschreibt, wie schnell enzymkatalysierte chemische Reaktionen verlaufen. Die Enzymkinetik findet breite Anwendung in Biologie und Medizin, da auch biologische Substrate (Reaktionspartner) – darunter solche, die im Menschen auftreten – untersucht werden. Ein Hauptziel der Enzymkinetik ist die Beschreibung der Konzentrationsabhängigkeit der Reaktionsgeschwindigkeit mit geeigneten Formeln, sowie die Bestimmung der dazugehörigen Parameter für ein bestimmtes Protein (Enzymaktivität und katalytische Effizienz). Da Enzyme dazu dienen, Reaktionen zu beschleunigen und zu lenken, ist die enzymkinetische Analyse zum Verständnis von Enzymfunktionen unerlässlich.
Theorie für Enzyme mit einer Substratbindungsstelle
BearbeitenDer Erste, der den Zusammenhang zwischen Substrat-Konzentration und Umsatzgeschwindigkeit eines Enzymes beschrieb, war der französische Physikochemiker Victor Henri 1902. Allerdings war die Bedeutung der Wasserstoffionenkonzentration für enzymatische Reaktionen damals noch nicht bekannt, erst nachdem Sørensen 1909 den pH-Wert definiert und die Pufferung eingeführt hatte, konnten der Deutsche Leonor Michaelis und seine kanadische Post-Doktorandin Maud Menten 1913 die Ergebnisse Henris experimentell bestätigen.[1] Die Henri-Michaelis-Menten-Gleichung wurde 1925 von G. E. Briggs und J. B. S. Haldane verallgemeinert (Michaelis-Menten-Theorie).
![{\displaystyle [S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/292bbb82029aa583c5d2ac5fa1d7e4fedf537d8b)

Henris Schlüsselidee war, die enzymatische Reaktion in zwei Phasen zu zerlegen, die Bindung des Substrates S an das Enzym E und die Umsetzung des resultierenden Enzym-Substrat-Komplexes ES in Enzym und Produkt P:
- (1)


Hierbei sind Geschwindigkeitskonstanten, die bei der kinetischen Herleitung des Massenwirkungsgesetzes (MWG) verwendet werden.[2] Zur Beschreibung eines Reaktionsgleichgewichts der Bindungsreaktion hat die Gleichheit der Geschwindigkeiten von Hin- und Rückreaktion die Form:

![{\displaystyle k_{1}[E][S]=k_{-1}[ES];\quad \mid :[ES]\quad \mid :k_{1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9c048c274c7fdd0fc05834b585f4b3da32f29386)
wobei die Konzentration der Substanz bezeichnet. Durch die angegebenen mathematischen Operationen entsteht für die Bindungsreaktion die eingeführte Formulierung des MWGs:
![{\displaystyle \textstyle [X]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d39b6a6cd2d0971206d3623299620f0720fe6318)

- (2)
![{\displaystyle {\frac {[E][S]}{[ES]}}={\frac {k_{-1}}{k_{1}}}=K_{\mathrm {d} }.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6b91e671eb7e9d727bd0b8fbf89f1b631491a548)
Da die (nach Standard im Zähler notierten) Reaktionsprodukte aus einer Dissoziation des Enzym-Substrat-Komplexes hervorgehen, wird die Gleichgewichtskonstante als Dissoziationskonstante bezeichnet.

Wie aus Gleichung (2) hervorgeht, hat die Dimension einer Konzentration. Für die Substratkonzentration ist die Hälfte aller Enzymmoleküle an Substrat gebunden, die andere Hälfte ist frei; dies wird als Halbsättigung des Enzyms bezeichnet. (Die Weiterreaktion bleibt zunächst außer Betracht.)
![{\displaystyle \textstyle [S]=K_{\mathrm {d} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/60ba695288cc922a10fdc204312e0c320603eac7)

| Rechnung hierzu |
Einsetzen von :
|
![{\displaystyle K_{\mathrm {d} }={\frac {[E][S]}{[ES]}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2359766ea2c2fe06edaf7e292e163d1b4a980b0d)
![{\displaystyle K_{\mathrm {d} }=[S]\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/562a12851b8ead62c71dc9f9b4ca194ba0de73cf)
![{\displaystyle [S]={\frac {[E][S]}{[ES]}};\quad \mid \cdot [ES]\quad \mid :[S]\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dd9cb42a0e81a26b1f6a5eaddc930d861098a529)
![{\displaystyle [ES]=[E]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a2b2e1aeb3db03a71353b882791593c54c27a3e6)
ist umgekehrt proportional zur Affinität des Enzymes für das Substrat: Je besser das Enzym das Substrat bindet, umso niedriger ist die für eine Halbsättigung des Enzyms erforderliche Substratkonzentration.
Zur Beschreibung eines Reaktionsgleichgewichts der Reaktion (1) insgesamt hat die Gleichheit der Geschwindigkeiten von Hin- und Rückreaktion die Form:
![{\displaystyle k_{1}[E][S]=k_{-1}[ES]+k_{cat}[ES]=(k_{-1}+k_{cat})[ES];\quad \mid :[ES]\quad \mid :k_{1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/aaef678468b25a2dfd0d3775d26027b3ba3dc1fa)
hierbei ist die Geschwindigkeitskonstante der (als nicht umkehrbar vorausgesetzten) Reaktion . Durch die angegebenen mathematischen Operationen entsteht für die Reaktion (1) die eingeführte Formulierung des MWGs:

- (3)
![{\displaystyle {\frac {[E][S]}{[ES]}}={\frac {k_{-1}+k_{cat}}{k_{1}}}=K_{m}.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/1c01543cc87063cae6ec3ec4d9687e9234285cdf)
heißt Michaelis-Menten-Konstante. Zur Beschreibung der Reaktionsgeschwindigkeit der betrachteten Katalyse wird (für entsprechend geeignete Fälle) weiter vorausgesetzt:

- Die Konzentration des insgesamt vorhandenen Enzyms ändert sich nicht und ist die Summe aus den Konzentrationen substratgebundenen und freien Enzyms, also .
- Die katalysierte Reaktion ist erster Ordnung, so dass ihre Geschwindigkeit zur Konzentration des Enzym-Substrat-Komplexes proportional ist, also .
- Eine maximale Reaktionsgeschwindigkeit wird als Rechengröße eingeführt. Diese entspricht dem fiktiven Fall, dass sämtliches vorhandenes Enzym als Enzym-Substrat-Komplex vorliegt, also .
![{\displaystyle [E]_{t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7a03490f35b5de1fef685c70b70e0569caddff4d)
![{\displaystyle [E]_{t}=[E]+[ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c3872a145753bee63b0ba1eb3d29cfa01d4e4ec7)
![{\displaystyle v=k_{cat}\cdot [ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5af917c21f01e7b0f5384f3eaa44bb2e3f21a33e)

![{\displaystyle v_{\mathrm {max} }=k_{cat}\cdot [E]_{t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3ae4b2fe049ba5e5729a6c80d7ca079a0c646e1e)
Durch Einführung dieser Bedingungen lässt sich (3) in die Michaelis-Menten-Gleichung umformen, die in Abhängigkeit von der Substratkonzentration darstellt:
![{\displaystyle v={\frac {v_{\mathrm {max} }\cdot [S]}{K_{m}+[S]}}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/52371624d9a314a09b138fa3f87e0352c32c3c16)
| Umformung |
|
Michaelis-Menten-Gleichung |
![{\displaystyle {\frac {v}{v_{\mathrm {max} }}}={\frac {k_{cat}\cdot [ES]}{k_{cat}\cdot [E]_{t}}}={\frac {[ES]}{[E]_{t}}}={\frac {[ES]}{[E]+[ES]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cbec5f44925a43c4326160772e34dead00610bb3)
![{\displaystyle \textstyle {\frac {[E][S]}{[ES]}}=K_{M}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/475fed22f595d48618e4b6752b0fca703d292703)
![{\displaystyle \textstyle {[S] \over [ES]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f4ce5ca0efed5141fd2178acbfc7c8f0b5552d02)
![{\displaystyle {\frac {v}{v_{\mathrm {max} }}}={\frac {[ES]}{[E]+[ES]}}={{[ES][S] \over [ES]} \over {[E][S] \over [ES]}+{[ES][S] \over [ES]}}\quad ={[S] \over K_{M}+[S]};\quad \mid \cdot v_{\mathrm {max} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/af7839618f41b52bedea7e960a8e90c400f7186b)
![{\displaystyle v={\frac {v_{\mathrm {max} }\cdot [S]}{K_{m}+[S]}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/afc1a0205643502364f4d5ef0ece783b9f8eff3a)
Der Graph dieser Gleichung ist Teil einer Hyperbel, die sich für zunehmende der waagerechten Asymptote nähert.

| Rechnung zur Klassifikation des Graphen als Teil einer Hyperbel; Betrachtung der Asymptoten |
|
A. Da und Konstanten sind, ist die Funktion eine Hyperbel mit der waagerechten Asymptote für und der senkrechten Asymptote für .
da durch eine Verkettung von Kongruenzabbildungen aus erzeugt werden kann, ist ebenfalls eine Hyperbel. Der Graph der Michaelis-Menton-Gleichung ist die Teilmenge von , für die ist. B. Die beiden zuerst genannten Kongruenzabbildungen ändern nichts an der waagerechten Asymptote der Hyperbel, die letztgenannte verschiebt sie um in -Richtung. Also hat die Asymptote für ; der in enthaltenen Graphen der Michaelis-Menton-Gleichung strebt für gegen diese Asymptote. Die zuerst genannte Kongruenzabbildungen verschiebt die senkrechte Asymptote der Hyperbel um in -Richtung, die beiden letztgenannten ändern nichts an ihr. Also hat die Asymptote für . |
![{\displaystyle v=f_{1}([S])={\frac {v_{\mathrm {max} }\cdot K_{m}}{[S]}};\quad [S]\in \mathbb {R} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/466109532c66dcbfb7600e4877f53315e796d2e2)

![{\displaystyle [S]\to \pm \infty }](https://wikimedia.org/api/rest_v1/media/math/render/svg/98f17f6e7a0400efe6e12ee83615c8af9e1c04cf)
![{\displaystyle [S]=0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d903cd589ae6f5ab9af262c90dcf5e25d9443529)
![{\displaystyle [S]\to 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5d459b85aecf5601e11194ab75099aacb5bfe827)

![{\displaystyle v=f_{2}([S])={\frac {v_{\mathrm {max} }\cdot K_{m}}{[S]-(-K_{m})}}={\frac {v_{\mathrm {max} }\cdot K_{m}}{K_{m}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/942957622a30956f79623b34e315e38cc8ef7bb0)
![{\displaystyle v=f_{3}([S])=-{\frac {v_{\mathrm {max} }\cdot K_{m}}{K_{m}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c817bf49e05b791da10d4e138e374fe0ad1c4740)

![{\displaystyle v=f_{4}([S])\quad =v_{\mathrm {max} }-{\frac {v_{\mathrm {max} }\cdot K_{m}}{K_{m}+[S]}}\quad =v_{\mathrm {max} }\cdot (1-{\frac {K_{m}}{K_{m}+[S]}})\quad =v_{\mathrm {max} }\cdot {\frac {K_{m}+[S]-K_{m}}{K_{m}+[S]}}\quad ={\frac {v_{\mathrm {max} }\cdot [S]}{K_{m}+[S]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/01869cc25123cfd2695f3d2dfb01c1c07d2201d3)
![{\displaystyle f_{4}([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f29a671dd8201841de9819bbb51e4e3d784c8d8)
![{\displaystyle f_{1}([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/558fa04cd75fa5fcf0673c6fb1a1657fdf79cc6e)
![{\displaystyle [S]\geq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9742d3fc03898a33597844707834ca2d2e592186)
![{\displaystyle [S]\to +\infty }](https://wikimedia.org/api/rest_v1/media/math/render/svg/3a6c5effaab5d83f890a4d62099c6f90cd9dba31)

![{\displaystyle [S]=-K_{M}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b7d8bedf2942bd388c9fec402e4c1cfaff126462)
![{\displaystyle [S]\to -K_{M}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/619f0c3d20d9d05056990720480ba071c94c7b1e)
Wie aus Gleichung (3) hervorgeht, hat auch die Dimension einer Konzentration. Für die Substratkonzentration ist .

![{\displaystyle \textstyle [S]=K_{m}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c5ee48c65e24e1ade6c7980b6f881ae2c4522d6b)

| Rechnung hierzu |
|
Einsetzen von in die Michaelis-Menton-Gleichung: . |
![{\displaystyle K_{m}=[S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/82041634b3d16cf053ae48a590b1498f99a69a7f)
![{\displaystyle \ v={v_{\mathrm {max} }\cdot [S] \over [S]+[S]}={v_{\mathrm {max} }\cdot [S] \over 2\cdot [S]}={\frac {v_{\mathrm {max} }}{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2db0a183d0e2b917cae600ee6dc09dfc4362edf8)
Zur Bestimmung von und aus Messreihen von und dienen computergestützte Verfahren wie die nichtlineare Regressionsanalyse (Simplex- oder Levenberg-Marquardt-Verfahren). Graphische Extrapolationsverfahren (Linearisierungen) wie etwa die doppelt-reziproke Auftragung nach Lineweaver und Burk sollten dafür nicht verwendet werden, da sie zu ungenau sind. Sie eignet sich jedoch sehr gut zur Präsentation der Ergebnisse enzymkinetischer Versuche.

Theorie für Enzyme mit mehreren Substratbindungsstellen
BearbeitenDie Hill-Gleichung und ihre Herleitung aus dem Massenwirkungsgesetz
BearbeitenDie Hill-Gleichung wurde ursprünglich von Archibald Vivian Hill eingeführt, um die Sauerstoffbindung an Hämoglobin in Abhängigkeit von verschiedenen Sauerstoffkonzentrationen mathematisch zu beschreiben.[3] Die hier beschriebene Hill-Gleichung ist eine andere als die Hill-Gleichung zur Beschreibung der Muskelkontraktion, an deren Erstellung der gleiche Autor beteiligt war.[4]
Obwohl die Bindung von Sauerstoff an Hämoglobin kein katalytischer Vorgang ist, lässt sich mit einer Hill-Gleichung auch die Kinetik enzymatischer Katalysen beschreiben, insbesondere auch solcher, deren Kinetik sich nicht mit einer Michaelis-Menten-Gleichung beschreiben lässt. Hier folgt eine Herleitung der Hill-Gleichung aus dem Massenwirkungsgesetz, die die Analogie zur Herleitung der Michaelis-Menten-Gleichung hervorhebt. Entsprechend bedeutet die Variable die Anzahl der Bindungsstellen, die ein Molekül Enzym für je ein Molekül Substrat bereithält, und ist damit eine positive natürliche Zahl. Die experimentell gefundenen Werte von weichen hiervon ab (s. u. "Der empirische Hill-Koeffizienten als Maß der Kooperativität von Enzymen").


Die Bindung von Molekülen Substrat an ein Enzym lässt sich modellieren mit:
- (1')

Wie in Gleichung (1) sind Geschwindigkeitskonstanten, die bei der kinetischen Herleitung des Massenwirkungsgesetzes (MWG) verwendet werden. Zur Beschreibung eines Reaktionsgleichgewichts der Bindungsreaktion hat die Gleichheit der Geschwindigkeiten von Hin- und Rückreaktion die Form:

![{\displaystyle k_{1}'[E][S]^{n}=k_{-1}'[ES_{n}];\quad \mid :[ES_{n}]\quad \mid :k_{1}'\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/feec7db7f954ece47a12a9287bc9a77c0b5e9e28)
hierbei ist die Konzentration freien Enzyms, die Substratkonzentration, die Konzentration der Enzym-Substrat-Komplexe mit Molekülen Substrat. Der Exponent heißt Hill-Koeffizient. Durch die angegebenen mathematischen Operationen entsteht für die Bindungsreaktion die eingeführte Formulierung des MWGs:
![{\displaystyle [E]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a170d18691c57fbfee5802ee401bd9f84ac8804b)
![{\displaystyle [ES_{n}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/75e237cdd690436419a2df4a52e9d23f67536962)
- (2')
![{\displaystyle {\frac {[E][S]^{n}}{[ES_{n}]}}={k_{-1}' \over k_{1}'}=K_{\mathrm {D} }.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/3ff3a899ee555eb8b6b8e9d1085c2dcd3d52ef09)
Analog der Dissoziationskonstante in Gleichung (2) heißt scheinbare Dissoziationskonstante. Das Adjektiv "scheinbar" trägt der Tatsache Rechnung, dass die experimentell gemessenen Werte für von den nach diesem Modell zu erwartenden abweichen.


Wie aus Gleichung (2') hervorgeht, hat die (neu einzuführende) Konstante
- (3')

die Dimension einer Konzentration. Für die Substratkonzentration ist die Hälfte aller Enzymmoleküle an Substrat gebunden, die andere Hälfte ist frei; dies wird als Halbsättigung des Enzyms bezeichnet.
![{\displaystyle \textstyle [S]=K_{A}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/be3623152c4e3dcc01c782e93217698097dd666d)
| Rechnung |
|
Gleichsetzen der Gleichungen (2') und (3') ergibt: Einsetzen von :
|
![{\displaystyle {\frac {[E][S]^{n}}{[ES_{n}]}}=\quad K_{\mathrm {D} }=\quad K{_{A}}^{n}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/48c52a0dbecc973233da852c9ed9667784cf5e57)
![{\displaystyle K_{A}=[S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2d2a0471ce0f32ec451b610485e47dc85def51d0)
![{\displaystyle [S]^{n}={\frac {[E][S]^{n}}{[ES_{n}]}};\quad \mid \cdot [ES_{n}]\quad \mid :[S]^{n}\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c57b75aad43522e966efe6b6c1b40043696b5a12)
![{\displaystyle [ES_{n}]=[E]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d0c3c34dce9f8048b5c0475c95da19547dc6fc74)
wird daher als Halbsättigungskonstante bezeichnet[5] und auch (für „50%“) geschrieben. ist (wie die Konstante der Michaelis-Menten-Gleichung) umgekehrt proportional zur Affinität des Enzymes für das Substrat: Je besser das Enzym das Substrat bindet, umso niedriger ist die für eine Halbsättigung des Enzyms erforderliche Substratkonzentration.



Wenn weiter vorausgesetzt wird,
- dass sich die Konzentration des insgesamt vorhandenen Enzyms nicht ändert und die Summe aus den Konzentrationen substratgebundenen und freien Enzyms ist, also ,
![{\displaystyle [E]_{t}=[E]+[ES_{n}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2ec199e4f3629c9b97041c1fd3ba547dc977483a)
dann ist der Anteil substratgebundenen Enzyms an insgesamt vorhandenem mit Gleichung (2'):

- Hill-Gleichung
![{\displaystyle \theta ={[ES_{n}] \over [E]_{t}}={[S]^{n} \over K_{D}+[S]^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d72a84a80f2f1d24db7dba113ed9b812e3689cf7)
| Rechnung |
|
Hill-Gleichung |
![{\displaystyle \theta ={[ES_{n}] \over [E]_{t}}={[ES_{n}] \over [E]+[ES_{n}]};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4c4e28b87e075d7ceecb3cf75f4fedc28d2e76a7)
![{\displaystyle \textstyle {\frac {[E][S]^{n}}{[ES_{n}]}}=K_{\mathrm {D} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4934d7aa04f4c8f88e6e4cbc7f74e73ab0d7f1b3)
![{\displaystyle \textstyle {[S]^{n} \over [ES_{n}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/24981f16cdaaf3e024180f0e75e821f9ab44f943)
![{\displaystyle \theta ={[ES_{n}] \over [E]+[ES_{n}]}={{[ES_{n}][S]^{n} \over [ES_{n}]} \over {[E][S]^{n} \over [ES_{n}]}+{[ES_{n}][S]^{n} \over [ES_{n}]}}\quad ={[S]^{n} \over K_{\mathrm {D} }+[S]^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/0d1e94ebbe5735d5d4f0e48c516303991d40194a)
Um mit der Hill-Gleichung die Reaktionsgeschwindigkeit der Katalyse durch ein Enzym mit mehreren Bindungsstellen zu beschreiben, ist hinreichend, weiter vorauszusetzen:
- Eine maximale Reaktionsgeschwindigkeit wird als Rechengröße eingeführt. Diese entspricht dem fiktiven Fall, dass sämtliches vorhandene Enzym als Enzym-Substrat-Komplex vorliegt, also .
- ist zum Anteil substratgebundenen Enzyms an insgesamt vorhandenem proportional.


Dann hat die Proportionalität die Form
- (4)

| Rechnung |
|
Wegen der vorausgesetzten Proportionalität von und zu existiert ein Proportionalitätsfaktor so, dass:
Damit ist das Verhältnis von zu :
|




Gleichsetzen mit der Hill-Gleichung ergibt eine Gleichung, die in Abhängigkeit von der -ten Potenz der Substratkonzentration darstellt:
![{\displaystyle [S]^{n}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a2baf5db0de3fbbf16e631569536f9f3032bd501)
- (5)
![{\displaystyle v={\frac {v_{\text{max}}\cdot [S]^{n}}{K_{D}+[S]^{n}}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/f7a0359434e2705d8dce82e9dfe1667c89d772da)
| Rechnung |
|
Gleichsetzen von Gleichung (4) mit der Hill-Gleichung ergibt: (5) |
![{\displaystyle {\frac {v}{v_{\text{max}}}}=\theta ={[S]^{n} \over K_{D}+[S]^{n}};\quad \mid \cdot v_{\text{max}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e588a78ee012bee7620c87b0ee4eee50cdcd2c1b)
![{\displaystyle v={v_{\text{max}}\cdot [S]^{n} \over K_{D}+[S]^{n}}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac85f4c0004624f80547b2a7345408b4cf47a739)
Die Herleitung der Gleichung (5) ist der Herleitung der Michaelis-Menten-Gleichung größtenteils analog. Unterschiede sind:
- Die Geschwindigkeitskonstante der katalysierten Reaktion wird nicht in die Herleitung von Gleichung (5) einbezogen: hängt im Gegensatz zu formal nicht von ab.
- Die Ordnung der katalysierten Reaktion wird bei der Herleitung von (5) nicht explizit betrachtet.

Statt der beiden letztgenannten Voraussetzungen geht die in Gleichung (4) formulierte Proportionalität in die Herleitung ein; ein abstrakter Proportionalitätsfaktor tritt an die Stelle von .
Weitere Darstellung für θ und für v. Die Sättigungsfunktion
BearbeitenIn der Hill-Gleichung ist von und von abhängig, selbst aber auch von (siehe Gleichung (2')). Das Verhalten der Gleichung in Abhängigkeit von ist einheitlicher darstellbar (s. u. halblogarithmisch aufgetragene Graphen), wenn durch ersetzt wird:
- (6)
![{\displaystyle \theta ={[S]^{n} \over K_{D}+[S]^{n}}={1 \over 1+({K_{A} \over [S]})^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/000be5773fdb4edbdd62872813a04937537c41a6)
| Umformung |
|
Einsetzen von (3'): in die Hill-Gleichung ergibt: mit erweitern und im Zähler und Nenner kürzen: (6) |

![{\displaystyle \theta ={[S]^{n} \over [S]^{n}+K{_{A}}^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d707627b84597fcf6b33da752ef81a8cb7dc4f84)
![{\displaystyle \textstyle {1 \over [S]^{n}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4a92536df12001a6ec34b5e429815adbf245b7d2)
![{\displaystyle \textstyle {[S]^{n} \over [S]^{n}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a2aeb2a4cc9449705055ad192f315a1bee91021f)
![{\displaystyle \theta ={1 \over 1+({K_{A} \over [S]})^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac6c9fa53b8a83f943bfc1a7a9b8eeed1ebe8988)
Gleichsetzen der Gleichungen (4) und (6) ergibt eine Darstellung von , die ebenfalls nicht mehr enthält:
- (7)
![{\displaystyle v={v_{\mathrm {max} } \over 1+({K_{A} \over [S]})^{n}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/f17ae68bd0f2b7ba2bab2f39f86e998e8500b442)
| Rechnung |
|
Gleichsetzen der Gleichungen (4) und (6) ergibt:
|
![{\displaystyle {v \over v_{\mathrm {max} }}=\theta ={1 \over 1+({K_{A} \over [S]})^{n}};\quad \mid \cdot v_{\mathrm {max} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/36c122e65341d4c3450d3fbd8c7aa87103d8bc62)
Wenn an ein Molekül Enzym Moleküle Substrat gebunden sind und die Konzentration der Enzym-Substrat-Komplexe ist, so ist die Konzentration des gebundenen Substrats . Als Sättigungsfunktion wird das Verhältnis der Konzentration gebundenen Substrats zur Konzentration des insgesamt vorhandenen Enzyms bezeichnet:[6]
![{\displaystyle n\cdot [ES_{n}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c95ee6de0eb15ab7842908b35591cecb9bb23d63)

![{\displaystyle r={n\cdot [ES_{n}] \over [E]_{t}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/695417a43b6e32f6421024fff217e70a7dd8a006)
Der Zusammenhang zur Hill-Gleichung ist wegen gegeben mit
![{\displaystyle \textstyle \theta ={[ES_{n}] \over [E]_{t}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f554f38fc6c3a05d2f4d5b161eee3e4f83da2ee1)
- (8)

Der empirische Hill-Koeffizienten nH als Maß der Kooperativität von Enzymen
BearbeitenGemäß Herleitung der Hill-Gleichung aus dem Massenwirkungsgesetz (s. o.) ist der Hill-Koeffizient die Anzahl der Bindungsstellen eines Enzyms und daher eine natürliche Zahl. (Genau) für sind die Konstanten und gleich. Auch sind genau für die Gleichungen (5) und (7) einer Michaelis-Menten-Gleichung äquivalent, indem die Konstante als Michaelis-Menten-Konstante aufgefasst wird.







| Rechnung für Gleichung (7) |
|
(7)
Michaelis-Menten-Gleichung |
![{\displaystyle v={v_{\mathrm {max} }\cdot [S] \over [S]+{K_{A}\cdot [S] \over [S]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5e910ab1d8fc4e1dd3c3551108ebb055ca2cadf6)

![{\displaystyle v={v_{\mathrm {max} }\cdot [S] \over [S]+K_{M}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/9a747fe1791ba985a80e10c6deb381ae83284670)
Zu Unterscheidung von wird mit der Variable derjenige Hill-Koeffizient bezeichnet, für den die Hill-Gleichung die Kinetik eines solchen Enzyms empirisch am besten beschreibt. ist in der Regel kleiner als und keine natürliche Zahl. Die Theorie der Hill-Gleichung ist bei Verwendung von nur dann mathematisch konsistent, wenn in allen zur Beschreibung der Kinetik verwendeten Gleichungen durch ersetzt wird. (9)

In Folgenden seien die Konstanten und in allen zu vergleichenden Situationen der jeweils betrachteten Enzyme gleich. Der Unterschied zwischen und wird dadurch erklärt, dass Enzyme mit mehreren Substratbindungsstellen aus mehreren Untereinheiten bestehen, die jeweils eine Bindungsstelle tragen und demzufolge für sich betrachtet mit und also einer Michaelis-Menten-Gleichung beschrieben werden können.


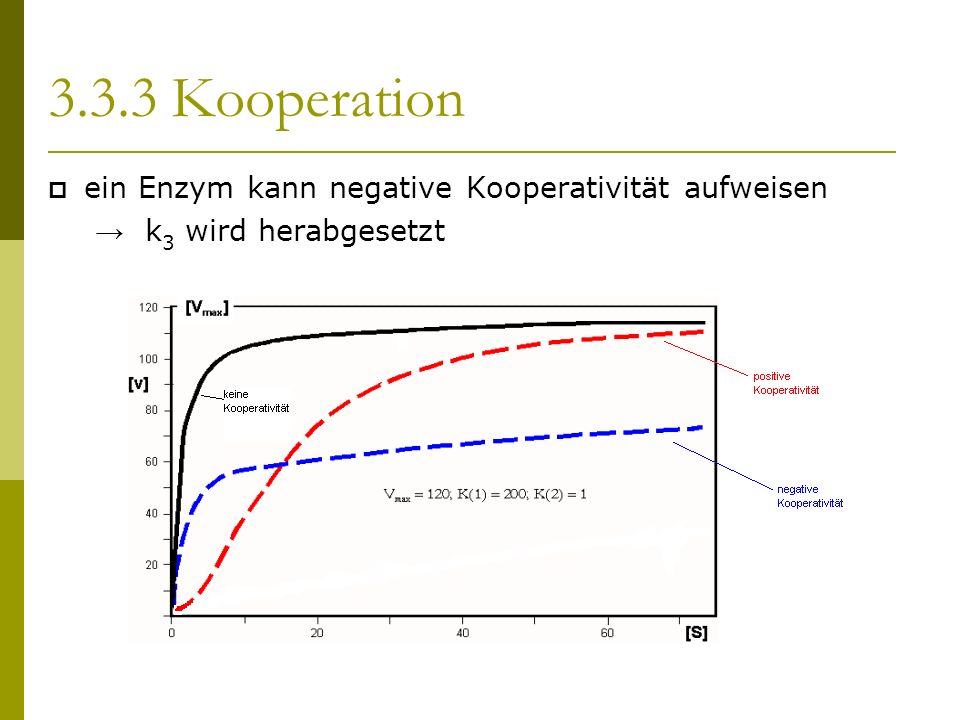
Ein als positive Kooperativität bezeichnetes Zusammenwirken der Untereinheiten kann aber auch bewirken, dass ein solches Enzym bei einer vorgegebenen Substratkonzentration schneller reagiert, als gemäß einer Michaelis-Menten-Gleichung (mit ) zu erwarten wäre. Eine Hill-Gleichung beschreibt für Konzentrationen genau dann positive Kooperativität, wenn ist. Weiter reagiert ein Enzym bei positiver Kooperativität bei einer vorgegebenen Substratkonzentrationen umso schneller, je größer ist. Logische Obergrenze für ist (die Anzahl der Bindungsstellen) .

![{\displaystyle [S]>K_{A}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/83eb5ceb369a9c05a0fd7c07138fd457e87f4575)

Ganz entsprechend kann ein als negative Kooperativität bezeichnetes Zusammenwirken von Untereinheiten eines Enzyms bewirken, dass jenes bei einer vorgegebenen Substratkonzentration langsamer reagiert, als gemäß einer Michaelis-Menten-Gleichung (mit ) zu erwarten wäre. Eine Hill-Gleichung beschreibt für Konzentrationen genau dann negative Kooperativität, wenn ist, und bei einer vorgegebenen Substratkonzentrationen reagiert ein Enzym bei negativer Kooperativität umso langsamer, je kleiner ist.

| Beweis |
|
Die folgende Ungleichung (i) verwendet Gleichung (7) zur Berechnung der Geschwindigkeiten zweier Enzyme, deren Situationen sich ausschließlich im Hill-Koeffizienten bzw. unterscheiden. Die folgenden Äquivalenzumformungen führen (i) auf die im Text genannten Bedingungen zurück.
der übersichtlicheren Schreibweise dient die Substitution . Nach (notwendiger) zusätzlicher Voraussetzung gilt
denn für die Überlegung kann vorausgesetzt werden. Einsetzen ergibt:
|

![{\displaystyle {v_{\mathrm {max} } \over 1+({K_{A} \over [S]})^{n_{H}}}>{v_{\mathrm {max} } \over 1+({K_{A} \over [S]})^{n{_{H}}'}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/477a3708ccfa725a4741096e2d6d0bfa8fd56659)
![{\displaystyle \textstyle q:={K_{A} \over [S]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6a538a84258b7a778da3bfcf895b6941b0949914)
![{\displaystyle [S]>K_{A}\quad \mid :[S]>0\Rightarrow }](https://wikimedia.org/api/rest_v1/media/math/render/svg/a968ac8b07fa5ad40aa04d07ba37e30381933d26)
![{\displaystyle 1>{K_{A} \over [S]}=q>0,\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/414fc0edcd28ef3777b2791e3f174a226740d8c0)
![{\displaystyle K_{A},[S]>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1fce220e9995bc1ce8bcd3d0a397b2944f5745b3)











Ein Enzym mit mehreren Bindungsstellen, bei dem ein solches Zusammenwirken der Untereinheiten nicht zu beobachten ist, heißt nicht kooperativ.
Kooperativität ist nicht nur für Enzyme beschrieben, sondern auch für Nicht-Enzym-Proteine, an die mehrere andere Moleküle binden (s. o. Herleitung der Hill-Gleichung). Für die koordinative Bindung von Sauerstoff an Hämoglobin, das aus je ein Sauerstoffmolekül bindenden Untereinheiten besteht, wurde ein Hill-Koeffizient von 2,8 bestimmt.[7]

Berechnung von nH
BearbeitenSind die Substratkonzentrationen bzw. bekannt, bei denen ein Enzym mit 10 % bzw. 90 % seiner Maximalgeschwindigkeit reagiert, so lässt sich sein empirischer Hill-Koeffizient bestimmen:
![{\displaystyle [S]=\mathrm {EC_{10}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/82536f160130678969cb6e6895ac307c125400ed)
![{\displaystyle [S]=\mathrm {EC_{90}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/65040e67635f7b3c620d289a431c8ecbf8149d0d)

Verallgemeinerung: Sind zwei beliebige verschiedene Substratkonzentrationen bzw. bekannt, bei denen ein Enzym mit 0%< P% <100% bzw. 0%< Q% <100% seiner Maximalgeschwindigkeit reagiert, so ist sein empirischer Hill-Koeffizient durch den folgenden Quotienten gegeben:



| Herleitung |
|
A. Mit Überlegung (9) ist bei Betrachtung des empirischen Hill-Koeffizienten in Gleichung (6) durch zu ersetzen. Die folgenden Umformungen lösen die entstehende Gleichung nach auf:
B. Mit Gleichung (4): ist außer durch die Hill-Gleichung auch durch den Anteil der gemessenen Reaktionsgeschwindigkeit an der Maximalgeschwindigkeit gegeben; dieser Anteil kann als Prozentsatz oder als Dezimalzahl angegeben sein. - Einsetzen von für bzw. von für in (i) ergibt:
C. (ii) und (iii) ergeben die Proportionalität: Mit einem Logarithmus zu einer wählbaren Basis und der Rechenregel für den Logarithmus einer Potenz:
D. Verallgemeinerung: Für zwei beliebige verschiedene Anteile bzw. von und den zugehörigen Substratkonzentrationen bzw. ergibt der gleiche Rechenweg:
wobei der Bruch im letzten Schritt mit erweitert wurde; mit Division durch den (nach Konstruktion von null verschiedenen) Faktor folgt die angegebene Formel. |
![{\displaystyle [S]^{n_{H}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/511f54150fa52d4b1ee1eeda3b2309bfda5cdac8)
![{\displaystyle \theta ={1 \over 1+({K_{A} \over [S]})^{n_{H}}};\quad \quad \mid }](https://wikimedia.org/api/rest_v1/media/math/render/svg/4f201ca74a3ef3173e5cb872913f35f58b81506a)

![{\displaystyle {1 \over \theta }-1={1-\theta \over \theta }={{K_{A}}^{n_{H}} \over [S]^{n_{H}}};\quad \mid }](https://wikimedia.org/api/rest_v1/media/math/render/svg/942a8bdc068149da4579b4c768c313372c568d4c)

![{\displaystyle {{K_{A}}^{n_{H}}\cdot \theta \over 1-\theta }=[S]^{n_{H}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fafedd7cdef2343cad83fe637fc31d2f82e79161)










![{\displaystyle [S]=EC_{P=100p}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/78c49c2e64fbf97b4df735d273e4142fdce5591e)
![{\displaystyle [S]=EC_{Q=100q}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9f69c64a9ec92b41546480ea3c571c484add5f97)





Nicht linearisierte Graphen
BearbeitenDirekt-lineare Auftragung einer Enzymkinetik nach Michaelis-Menten
Bearbeiten
Enzymkinetische Parameter lassen sich bequem und präzise direkt aus einer Sättigungshyperbel gemäß der Abbildung herleiten („direkt-lineare Auftragung“ auch „Cornish-Bowden-Diagramm“ genannt). In dieser Hyperbel ist die enzymatische Umsatzgeschwindigkeit (Ordinate) als Funktion der Substratkonzentration (Abszisse) dargestellt.
Für die direkt-lineare Auftragung überträgt man die Anfangsgeschwindigkeiten des enzymatischen Umsatzes direkt in das - -Diagramm. Die -Werte sind vor Versuchsbeginn bekannt (eingestellte Substratkonzentrationen); während der Versuchsreihe ist dann der Ordinatenwert für (die Anfangsgeschwindigkeit) nachzutragen. Aus der maximalen Umsatzgeschwindigkeit lässt sich die halbe maximale Umsatzgeschwindigkeit ableiten. Graphisch kann man daraus den Koordinatenwert für ermitteln. Die katalytische Effizienz folgt übrigens aus der Steigung der Tangente an den Ursprung: ; daraus ergibt sich .



| Berechnung der Steigung der Tangente an den Ursprung |
|
Die Funktionsgleichung der Hyperbel ist die Michaelis-Menten-Gleichung
die Steigung der Tangente an den Ursprung kann als Grenzwert einer Sekantensteigung aufgefasst werden, die durch einen Differenzenquotienten gegeben ist. Das ergibt bei Näherung von rechts:
|
![{\displaystyle v([S])=\quad {\frac {v_{\mathrm {max} }[S]}{K_{\mathrm {m} }+[S]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f0f12bea4609a473d80d8b7c2bf42e3d443ad5bb)





![{\displaystyle v([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4fb1cdb881b31e0df5e5a736b6708bc1fcb2aa85)
Die Fehlerbehandlung wird im direkt-linearen Plot weitgehend vereinfacht: Mittelwertsbildung gibt dann die wahrscheinlichen Werte für die Parameter und . Bei Inspektion der Streubreite der Messpunkte (nicht identisch mit deren Standardabweichung) können Ausreißer leicht identifiziert und sogenannte Mediane abgelesen werden.
An dieser Stelle sei erwähnt, dass alle (auch die nachfolgenden) Auswertungsverfahren nicht nur für Enzyme, sondern auch für die Bindungsvorgänge von Carriern oder Rezeptoren Gültigkeit haben. Historisch gesehen wurden all diese Methoden (Hanes und Eadie-Hofstee-Auftragung für Enzyme, Scatchard und Hill-Auftragungen für Carrier) ursprünglich von Woolf entwickelt.
Direkt-linear aufgetragene Graphen einer Enzymkinetik nach Hill für unterschiedliche Werte von nH
BearbeitenDie aus der Hill-Gleichung hergeleitete Gleichung (5) lässt sich als eine Funktion auffassen, die die empirisch gefundene Reaktionsgeschwindigkeit abhängig von der Substratkonzentration beschreibt. Nach Überlegung (9) ist bei der Formulierung der Funktion durch zu ersetzen:
![{\displaystyle f([S])=v={v_{\mathrm {max} }\cdot [S]^{n_{H}} \over K_{D}+[S]^{n_{H}}}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7df3f7891d849c3b8b8a3583dd23463115fd9ac3)
f([S]) ist überall streng monoton steigend und nähert sich für zunehmende der waagerechten Asymptote . Der Graph von f([S]) zeigt aber je nach Wert von unterschiedliches Verhalten:[8]
- Für ist er Teil einer Hyperbel, da Gleichung (5) genau dann einer Michaelis-Menten-Gleichung äquivalent ist (s. o.).
- Für hat er genau einen Wendepunkt bei . Unter Mitberücksichtigung ihres Steigungsverhaltens ist daher in diesem Fall eine Sigmoidfunktion. Der Fall lässt sich von den Fällen und durch bloße Betrachtung des Funktionsgraphen unterscheiden.
- Für hat er keinen Wendepunkt und sieht einem Teil einer Hyperbel ähnlich. Ein solcher Graph heißt pseudohyperbol, weil sich der Fall vom Fall durch bloße Betrachtung des Graphen nicht unterscheiden lässt.
![{\displaystyle \textstyle [S]_{w}={\Big (}K_{D}\cdot {n_{H}-1 \over n_{H}+1}{\Big )}^{1 \over n_{H}}=K_{A}\cdot {\Big (}{n_{H}-1 \over n_{H}+1}{\Big )}^{1 \over n_{H}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ed804abe06764151e428b607c1d0ff8a26a34aab)
![{\displaystyle f([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8c71ccaa1038293d21be468bc10db28072033012)
| Rechnerische Nachweise |
|
Vorüberlegungen:
A. Für beliebige ergibt die Konvergenz der Potenzfunktion:
B. Die Ableitung ist mit (i) und (iii) überall positiv für , sodass für alle streng monoton steigt. Zusätzliche Berücksichtigung von und zeigt, dass im gesamten Definitionsbereich streng monoton steigt, wie behauptet. C. Die Wendepunkte von sind genau die Nullstellen mit Vorzeichenwechsel der zweiten Ableitung
Da für nicht definiert ist, wechselt in nicht das Vorzeichen, und hat bei keinen Wendepunkt. Außer gilt für alle Wendepunkte:
Mit (ii) hat Gleichung (iv) genau dann eine Lösung, wenn ist, und dann genau eine (d. h. ihre Lösung ist eindeutig, falls sie existiert). Mit ist für , sodass keine Lösung von (iv) existiert und keinen Wendepunkt hat, wie behauptet. Mit ist für , sodass dann genau eine Lösung von Gleichung (iv) existiert; hat bei nach (ii) höchstens einen Wendepunkt. Zu zeigen bleibt, dass in das Vorzeichen wechselt. Da die Bestimmung der dritten Ableitung recht aufwendig ist, wird hier das Verhalten von in einer Umgebung ihrer Nullstellen untersucht. Für beliebige sind Potenzfunktionen streng monoton steigend. Also gilt für beliebige , für die ist:
Für eine geeignete -Umgebung von ist also für alle (v) Mit zeigen ausgehend von die entsprechenden Umformungen, dass für alle ist. Letzteres zeigt zusammen mit (v) den Vorzeichenwechsel von in . |


![{\displaystyle [S]\rightarrow [S]^{a}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/256e8e6b306e374943e13dd175ae8d86de0d58e5)
![{\displaystyle [S]>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9dda6d969ffb31cf31b9ba8f29e66ed7a676b2d3)

![{\displaystyle K_{D},[S]>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1bdc0cd75e76332aa56c9ad603c1e6a371837017)
![{\displaystyle [S]^{n_{H}},[S]^{n_{H}-1},[S]^{n_{H}-2},(K_{D}+[S]^{n_{H}})^{2},(K_{D}+[S]^{n_{H}})^{3}>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4178ca96b07d28329dc319c3e06149f8a4869d04)

![{\displaystyle \lim _{[S]\to \infty }{K_{D} \over [S]^{n_{H}}}=K_{D}\cdot \lim _{[S]\to \infty }[S]^{-n_{H}}=K_{D}\cdot 0=0;\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/8c29e5449ba8257f654662354918d562825dc5db)
![{\displaystyle \textstyle f([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9202925d108b5721f9a6ed60c94032d7011508eb)
![{\displaystyle \textstyle {1 \over [S]^{n_{H}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5d93f3555c95d3f2cd8bf6a7401eada76027cbad)
![{\displaystyle \lim _{[S]\to \infty }{v_{\mathrm {max} }\cdot [S]^{n_{H}} \over K_{D}+[S]^{n_{H}}}=\lim _{[S]\to \infty }{v_{\mathrm {max} }\cdot {[S]^{n_{H}} \over [S]^{n_{H}}} \over {K_{D} \over [S]^{n_{H}}}+{[S]^{n_{H}} \over [S]^{n_{H}}}}={v_{\mathrm {max} }\cdot 1 \over 0+1}=v_{\mathrm {max} },\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/5bcb1822ab735f23b7246cf4a8ead7d01da51f77)
![{\displaystyle f'([S])={v_{\mathrm {max} }\cdot K_{D}\cdot n_{H}\cdot [S]^{n_{H}-1} \over (K_{D}+[S]^{n_{H}})^{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac36a4f4bd48cbd24733421cbec60a6f69db68e7)

![{\displaystyle f([S]>0)>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9b2f74454acfc9797bbb514c65dd8ea4fae9ffda)
![{\displaystyle f''([S])=v_{\mathrm {max} }\cdot K_{D}\cdot n_{H}\cdot [S]^{n_{H}-2}\cdot {K_{D}(n_{H}-1)-[S]^{n{_{H}}}(n_{H}+1) \over (K_{D}+[S]^{n_{H}})^{3}}=a([S])\cdot {b([S]) \over c([S])},\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/64fdf9874e60f89ba6999d38340a506ed65f4248)
![{\displaystyle a([S]),b([S]),c([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/614f18199dd89d7ff2cac3e7f9e23c7f715204ad)
![{\displaystyle a([S])=v_{\mathrm {max} }\cdot K_{D}\cdot n_{H}\cdot [S]^{n_{H}-2}>0\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d47975af5ceda9c82c48450eebf3b5b4bf8ead92)
![{\displaystyle \quad [S]>0\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/38f727c287b5921846f9c8947694a183bf55a5cc)
![{\displaystyle b([S])=K_{D}(n_{H}-1)-[S]^{n{_{H}}}(n_{H}+1)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/edf95c6bcc96043b34f40dfdeda872deee28127b)
![{\displaystyle c([S])=(K_{D}+[S]^{n_{H}})^{3}>0\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6eee8a34c284160eae48811a4ef50e90385c84a8)
![{\displaystyle f''([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/130f2e0a29a0717aabc18334790e337612762be4)
![{\displaystyle [S]<0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a0ff6133c00551f115bc6473cb8eaade994f780)
![{\displaystyle f''([S])=0\quad \Leftrightarrow \quad b([S])=0\quad \Leftrightarrow \quad [S]^{n_{H}}=K_{D}\cdot {n_{H}-1 \over n_{H}+1}=:D.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fc1af1baa45802065d6b997030c5a7f34a24928b)

![{\displaystyle [S]=[S]_{w}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/368e50516b8a1f2e073ea605a8e61a1940c7a229)


![{\displaystyle [S]_{w}=D^{1 \over n_{H}}={\Big (}K_{D}\cdot {n_{H}-1 \over n_{H}+1}{\Big )}^{1 \over n_{H}}=K_{A}\cdot {\Big (}{n_{H}-1 \over n_{H}+1}{\Big )}^{1 \over n_{H}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/42c30fe7b36cc6d3b59ebd14121f8420d4cf0292)
![{\displaystyle [S]_{w}>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8448b454c5f00835086bc571a7f1e9c9e7b90f78)
![{\displaystyle f'''([S])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cfbb9a37256c8865ec1ff2b1c90a0eac23f08afa)
![{\displaystyle [S]_{w}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0566b2a3c6f366e00e59e7326094c48eaff8e95b)



![{\displaystyle [S]_{w}>\epsilon >0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/560dcbbe2b80980625671c0254ad6b9b6e2d8ce8)
![{\displaystyle ([S]_{w}-\epsilon )^{n{_{H}}}<{[S]_{w}}^{n{_{H}}}\quad \mid \cdot (-(n_{H}+1))<0\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/c26515beed5f68eaade92d55cf185a444c22ad9a)

![{\displaystyle -([S]_{w}-\epsilon )^{n{_{H}}}(n_{H}+1)>-{[S]_{w}}^{n{_{H}}}(n_{H}+1)\quad \mid +K_{D}(n_{H}-1)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5b43c76c3fa3fb3da18f3d23aa552bfd7a164da0)
![{\displaystyle b([S]_{w}-\epsilon )>b([S]_{w})=0\quad \mid \cdot a([S]_{w}-\epsilon )>0\quad \mid :c([S]_{w}-\epsilon )>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3d271b5766b5be878c40834367ff9e11ad6c73da)
![{\displaystyle f''([S]_{w}-\epsilon )>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b8106b3239f1d22d4c46cebd39143d26fe0921e6)
![{\displaystyle f''([S])>0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ddf2013511e253e0ddf6b898dd7842748099c90f)
![{\displaystyle [S]\in U_{\epsilon }([S]_{w}),\quad [S]<[S]_{w}.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/4a7754995a728ed52bb025c7a5fe2fa18a68e0f3)
![{\displaystyle [S]_{w}+\epsilon >0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f5f08fe3d62b21e4fa2c311d94764953200ba8f2)
![{\displaystyle ([S]_{w}+\epsilon )^{n{_{H}}}>[S_{w}]^{n{_{H}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/97734e73cf032b00f34902834bd1a44d74d0b389)
![{\displaystyle f''([S])<0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/258874a5f29d6e68291518fba0fd59bdaaff80a2)
![{\displaystyle [S]\in U_{\epsilon }([S]_{w}),\quad S>[S]_{w}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/82c6cdfbeba1c2060d567c4be117dee8e72b4ef3)
Halblogarithmisch aufgetragene Graphen einer Hill-Gleichung für unterschiedliche Werte von nH
Bearbeiten
![{\displaystyle \textstyle {[S] \over K_{A}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f7fb74225330a0a4e2d447c1cb2b6d9d6a62361d)
Im nebenstehenden Diagramm[9] ist die Ordinate der Anteil substratgebundenen Enzyms an insgesamt vorhandenem. Die Abszisse gibt das Verhältnis an; sie ist logarithmisch geteilt. Bei Verwendung des dekadischen Logarithmus ist
- wegen der Punkt „1“ Nullpunkt der Abszisse und
- wegen und der Abstand zwischen den Punkten „1“ und „10“ Längeneinheit der Abszisse.


Für beliebiges bezeichnet ein Längeneinheiten vom Nullpunkt entfernter Punkt der Abszisse die Substratkonzentration , wobei der Faktor auf der Abszisse ablesbar ist. Jeder Graph des Diagramms zeigt eine Hill-Gleichung der Form (6):


![{\displaystyle [S]=10^{x}\cdot K_{A}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e248e639bdd1575460141c05be726842dca30170)

- .
![{\displaystyle \theta ={1 \over 1+({K_{A} \over [S]})^{n_{H}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f97fc3d13dd3437a693fd710f1376efd38d39658)
| Erläuterung |
|
Jeder Graph des Diagramms zeigt eine Funktion
wie durch Betrachtung des Verhaltens dieser Funktion für (am Nullpunkt "1" der Abszisse) und für anschaulich wird. Einsetzen von zeigt, dass jeder Graph auch eine Hill-Gleichung der Form (6) darstellt:
|



![{\displaystyle (\quad [S]=10^{x}\cdot K_{A}\Leftrightarrow \quad {[S] \over K_{A}}=10^{x}\Leftrightarrow \quad )\quad {K_{A} \over [S]}=10^{-x}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/91f87fc94c84a1e9414d599930f7b7208ae94330)
![{\displaystyle \theta ={1 \over 1+(10^{-x})^{n_{H}}}={1 \over 1+({K_{A} \over [S]})^{n_{H}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9b809840833f8e8c2531aefe05ffea4327f0687d)
Für je einen vorgegebene Hill-Koeffizienten ist im Diagramm der Graph der Hill-Gleichungen zu allen Werten von gleich, da nicht direkt von abhängt, sondern vom Verhältnis . Die Gesamtheit der Graphen bildet für beliebige positive eine (einparametrige) Schar direkt vergleichbarer Kurven; Teile einer Hyperbel oder pseudohyperbole Kurvenverläufe treten nicht auf.

![{\displaystyle \textstyle {K_{A} \over [S]}(=10^{-x})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/595ef9960bc7376871bb1b17a7c34a3853be59fc)

Jeder Graph des Diagramms ist auch Graph einer logistischen Funktion und hat daher

- für die Asymptote sowie
- für die Asymptote ;
- jeder Graph ist punktsymmetrisch mit Symmetriezentrum ;
- ist der Wendepunkt jeder Funktion ;
- die Steigung von in ist . In diesem Sinne nimmt die Steilheit des Graphen mit zu.
![{\displaystyle x\rightarrow \infty \quad (\Leftrightarrow [S]\rightarrow \infty )\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/1cee259c661d24c0c5776397fce8c2db70b6ca88)
![{\displaystyle x\rightarrow -\infty \quad (\Leftrightarrow [S]\rightarrow 0)\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/5a463899ee8ca32d92c12afb6d658bddf92b87d9)





| Beweis |
|
Die allgemeine logistische Funktion kann geschrieben werden: ; mit hat die Form: (6), q. e. d. Die Eigenschaften der allgemeinen logistischen Funktion setzen sich auf wie folgt fort:
|


![{\displaystyle \quad G=1,\quad c=0,\quad k=n_{H}\cdot \ln(10),\quad [S]=10^{x}\cdot K_{A}\Leftrightarrow \quad {K_{A} \over [S]}=10^{-x}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/8f85fd1be9404d6db406b6cd8063156283cff77f)
![{\displaystyle \theta ={1 \over 1+e^{0}\cdot e^{-\ln(10)\cdot n_{H}\cdot 1\cdot x}}=\quad {1 \over 1+1\cdot 10^{-n_{H}\cdot x}}=\quad {1 \over 1+(10^{-x})^{n_{H}}}\quad ={1 \over 1+({K_{A} \over [S]})^{n_{H}}}\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/b8669d63924465869bcbe23f1d5075baaabe2f43)
![{\displaystyle x\rightarrow \infty \quad (\Leftrightarrow 10^{x}\cdot K_{A}=[S]\rightarrow \infty )}](https://wikimedia.org/api/rest_v1/media/math/render/svg/379e2e9d2f45a226334cc3341899c70652f2e4d3)

![{\displaystyle x\rightarrow -\infty \quad (\Leftrightarrow 10^{x}\cdot K_{A}=[S]\rightarrow 0)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d4c66df3b08c38bedd876b0f21f37bf52ffd3c1a)





Weiter gehört jede logistische Funktion zu den Sigmoidfunktionen, d. h. jeder ihrer Graphen verläuft S-förmig.
Linearisierungsverfahren
BearbeitenLinearisierungsverfahren wurden in der Vergangenheit sehr häufig für die schnelle grafische Bestimmung der wichtigen Kinetikparameter und verwendet. Sie sind zwar einprägsam und verbreitet, führen jedoch zu einer teils erheblichen Verfälschung des Ergebnisses durch Messfehler und sind zur Fehlerbetrachtung mehr oder weniger ungeeignet. Mittlerweile hat die Ermittlung der Michaelis-Menten-Parameter durch nichtlineare Regression stark an Bedeutung gewonnen, die zu deutlich genaueren Ergebnissen führt. Deshalb sollen die Linearisierungsverfahren hier nur gestreift werden.
Lineweaver-Burk-Diagramm
Bearbeiten
Hans Lineweaver (1907–2009) und Dean Burk (1904–1988) haben 1934 eine doppelt-reziproke Darstellung vorgestellt, bei der als Funktion von aufgetragen wird.[10]

![{\displaystyle {\tfrac {1}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/510219bf46e867abe73910afb38bedf1e52a7183)
Eine Umformung der Michaelis-Menten-Gleichung ergibt die folgende Gleichung:
![{\displaystyle {1 \over v}={K_{m} \over v_{\text{max}}}{1 \over [S]}+{1 \over v_{\text{max}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4faf30a04dd6a31cd3a684e0bb31cd49aeabf100)
| Umformung |
|
(Michaelis-Menten) Übergang zur reziproken Zahl
(Lineweaver-Burk) |
![{\displaystyle v=\quad {\frac {v_{\mathrm {max} }[S]}{K_{\mathrm {m} }+[S]}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/cd4bf5bb736605533318c7a04aa0e976e4932eb4)

![{\displaystyle {\frac {1}{v}}=\quad {\frac {K_{\mathrm {m} }+[S]}{v_{\mathrm {max} }[S]}}=\quad {\frac {K_{\mathrm {m} }}{v_{\mathrm {max} }[S]}}+{\frac {[S]}{v_{\mathrm {max} }[S]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f3fa2b707cf2ddf5a0b7fdaf253d5b1bf2a8cc36)
![{\displaystyle {1 \over v}={K_{m} \over v_{\text{max}}}{1 \over [S]}+{1 \over v_{\text{max}}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2ff78fdfe3bcc1af2266b5e3032a5866ba3a782b)
Die Steigung dieser linearen Funktion beträgt ; sie schneidet

- die -Achse bei (Ordinatenabschnitt) und
- die -Achse bei (Abszissenabschnitt).


![{\displaystyle \textstyle {1 \over [S]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9ce8552c34ca3d76c4d9448f6dadf696db0050f3)
![{\displaystyle \textstyle {1 \over [S]}=-{\tfrac {1}{K_{m}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4906da8e4c4426a96065b3c67bd5f8b891b0b1b6)
| Rechnung zu Achsenabschnitten und Steigung |
|
Indem als Argument einer Funktion aufgefasst wird, beschreibt die Gleichung eine lineare Funktion. Für diese lassen sich Steigung und Ordinatenabschnitt direkt aus der Funktionsgleichung ablesen. Sei
Dann ist das Dreieck einem Steigungsdreieck ähnlich. Also gilt für das Verhältnis der Streckenlängen von Ordinaten- und Abszissenabschnitt:
|



![{\displaystyle \textstyle {1 \over [v]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/11b5a525256d19e40e838af441efc58882b3713a)


![{\displaystyle \textstyle {\frac {1}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0e0fdf99d5ba5dd7fae9bf8f3483d27f1a4a5be6)







Obwohl sie zur Datenrepräsentation meist verwendet wird, ist diese Methode zur Auswertung jedoch unverlässlich. Kleine Fehler in ergeben bei kleinen -Werten eine große Abweichung in , bei großen -Werten ist diese eher zu vernachlässigen. Die Autoren der Methode haben die Unsicherheit großer Werte betont und darauf hingewiesen, dass diese grundsätzlich geringer zu gewichten sind. Spätere Anwender haben dies zumeist ignoriert. Wo immer möglich sollte dieses durch Computerverfahren zur Bestimmung enzymkinetischer Parameter ersetzt werden.
Eadie-Hofstee-Diagramm
BearbeitenDas Eadie-Hofstee-Diagramm, auch Woolf–Eadie–Augustinsson–Hofstee- oder Eadie–Augustinsson-Diagramm, nimmt eine Mittelstellung ein. Hierbei wird als Funktion von aufgefasst. Die zugehörige Umformung der Michaelis-Menten-Gleichung ergibt:
![{\displaystyle {\tfrac {v}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/74c98f65d286f614677d7bac0d99f7f8db65d592)
![{\displaystyle v=-K_{m}{v \over [S]}+v_{\text{max}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/025725a9469594533972d3bd5a400f27737f69f1)
| Umformung |
|
(Michaelis-Menten)
Umformung der linken Seite: einsetzen:
(Eadie-Hofstee) |
![{\displaystyle v=\quad v_{\mathrm {max} }\cdot {\frac {[S]}{K_{\mathrm {m} }+[S]}};\quad \mid \cdot {\frac {K_{\mathrm {m} }+[S]}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bd0ed55bad37c1cb3db53b771c622bd77eb20ebe)
![{\displaystyle v\cdot {\frac {K_{\mathrm {m} }+[S]}{[S]}}=v_{\mathrm {max} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f65a29b9240d4304f837ec3cbb7ddc7d8d041a37)
![{\displaystyle \quad v\cdot {\frac {K_{\mathrm {m} }+[S]}{[S]}}=\quad v\cdot ({\frac {K_{\mathrm {m} }}{[S]}}+1)=\quad v\cdot {\frac {K_{\mathrm {m} }}{[S]}}+v;\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/0fdf6b275f0a047fea441148e792131aaa119b8f)
![{\displaystyle v\cdot {\frac {K_{\mathrm {m} }}{[S]}}+v=v_{\mathrm {max} }\quad \mid -{\frac {v\cdot K_{\mathrm {m} }}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7d299130f32667abe701b9a267854f8c95d55b29)
![{\displaystyle v=-K_{\mathrm {m} }\cdot {\frac {v}{[S]}}+v_{\mathrm {max} };\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fee4853d0809a440e9dfa0dacbd8111ec9746787)
Aus dem Diagramm lässt sich auf der -Achse als Ordinatenabschnitt ablesen, aus der (negativen) Steigung der Regressionsgeraden bestimmen.
Der Fehler wächst mit v/[S]. Da v bei beiden Koordinaten eingeht, konvergieren alle Abweichungen zum Ursprung.
Scatchard-Diagramm
BearbeitenDas Scatchard-Diagramm fasst umgekehrt als Funktion von auf. Es entsteht aus dem Eadie-Hofstee-Diagramm durch Vertauschung der Achsen (oder äquivalent: durch Spiegelung des Diagramms insgesamt an der 1. Winkelhalbierenden des Koordinatensystems). Die entsprechende Umformung der zum Eadie-Hofstee-Diagramm gehörigen Gleichung ergibt:
![{\displaystyle v/[S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a302c215e47f8a733641e8c68c519d5644ebf701)
![{\displaystyle {\frac {v}{[S]}}=-{\frac {1}{K_{m}}}\cdot v+{\frac {v_{\text{max}}}{K_{m}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/eb719a7ed4698093cf9d6cb2da8b3adfbc0a200e)
| Umformung |
|
(Eadie-Hofstee)
(Scatchard) |
![{\displaystyle v=-K_{m}{v \over [S]}+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/dc7de6ec59d5ef4b3f0201b0cefbb06603ead08a)



![{\displaystyle -K_{m}{v \over [S]};\quad \mid :(-K_{m})\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b2edf9494e3073825c86ca96cb5510b49252c18a)
![{\displaystyle {v-v_{\text{max}} \over -K_{m}}=\quad {v_{\text{max}}-v \over K_{m}}=\quad {v \over [S]};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95e148e413e457fd7837eb36ddb6dc8f34d34b6d)
![{\displaystyle {v \over [S]}=-{v \over K_{m}}+{v_{\text{max}} \over K_{m}}=\quad -{1 \over K_{m}}\cdot v+{v_{\text{max}} \over K_{m}}.\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/c74215e762399cfcb4ee525d0cfa1031bffd58e0)
Aus dem Diagramm lässt sich ebenfalls auf der -Achse, die nun Abszisse ist, als Abszissenabschnitt ablesen, denn ein Ordinatenabschnitt des Eadie-Hofstee-Diagramms geht durch die genannte Spiegelung in einen Abszissenabschnitt des Scatchard-Diagramms über. Ebenso lässt sich aus der (negativen) Steigung der Regressionsgeraden durch Übergang zur reziproken Zahl und Vorzeichenwechsel bestimmen. Der Ordinatenabschnitt der Gerade im Scatchard-Diagramm ist der im Abschnitt "Direkt-lineare Auftragung" als Maß der katalytischen Effizienz genannte Bruch.

Das Scatchard-Diagramm wird zumeist zur Repräsentation von Bindungsmessungen (anstelle enzymkinetischer Daten) angewendet. Scatchard- und Eadie-Hofstee-Diagramme gelten als die besten Werkzeuge zur Diagnose kooperativer Phänomene. Im Falle negativer Kooperativität oder nicht-identischer, isolierter Bindungsplätze entsteht ein konkaver Verlauf mit linearem Endast. Die Steigungen entsprechen hier den Affinitäten (Kd beziehungsweise ) und die Gesamtzahl der Bindungsplätze (aktiven Zentren) ist aus dem Schnittpunkt mit der -Achse abzulesen.
Hanes-Woolf-Diagramm (Hanes(-Wilkinson)-Diagramm)
Bearbeiten
Das Hanes-Woolf-Diagramm[11] ist die bestmögliche lineare Auftragungsmöglichkeit. Es geht auf Charles Samuel Hanes (1903–1990) und Barnet Woolf (1902–1983) zurück. Hierbei wird eine Umformung der Michaelis-Menten-Gleichung verwendet, die [S]/v als Funktion von [S] darstellt:
![{\displaystyle {[S] \over v}={1 \over v_{\text{max}}}[S]+{K_{m} \over v_{\text{max}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8dbc70a39461fa19d05969019a30ade5fec216ea)
| Umformung |
|
(Michaelis-Menten) Brüche stürzen
(Hanes-Woolf) |
![{\displaystyle v=\quad {\frac {v_{\mathrm {max} }[S]}{K_{\mathrm {m} }+[S]}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fb0cb0ab1209b921760ca9ccad66ddc87270bb0c)
![{\displaystyle \quad \mid :[S]\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8623bef0442b1b4171282a47f5134c296d4862a3)
![{\displaystyle {\frac {[S]}{v}}=\quad {\frac {K_{\mathrm {m} }+[S]}{v_{\mathrm {max} }}}=\quad {\frac {[S]}{v_{\mathrm {max} }}}+{\frac {K_{\mathrm {m} }}{v_{\mathrm {max} }}};}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e1dfc2049883e98daa36f6185764cc4f8ff12589)
![{\displaystyle {\frac {[S]}{v}}=\quad {\frac {1}{v_{\text{max}}}}\cdot [S]+{\frac {K_{\mathrm {m} }}{v_{\text{max}}}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/56666a47461f1895ec4d7225274a434b489addac)
Die Steigung dieser linearen Funktion beträgt ; sie schneidet

- die -Achse bei (Ordinatenabschnitt) und
- die -Achse bei (Abszissenabschnitt).
![{\displaystyle \textstyle {\frac {[S]}{v}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/16e7301d366c124e37c8359f65df1f4af5a7147b)
![{\displaystyle \textstyle {\frac {[S]}{v}}={\frac {K_{\mathrm {m} }}{v_{\mathrm {max} }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/253a6b3bf61eac18bd2e6a6096239bc8e6057acc)
![{\displaystyle \textstyle [S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9f1dc724cb20a4823ff8982b6727b4f18dcc610d)
![{\displaystyle \textstyle [S]=-K_{\mathrm {m} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a118e8f8c59bf1710d772d32f177a16a4c22e704)
| Rechnung zu Achsenabschnitten und Steigung |
|
Für die lineare Funktion mit der Gleichung lassen sich Steigung und Ordinatenabschnitt direkt aus der Funktionsgleichung ablesen. Sei
Dann ist das Dreieck einem Steigungsdreieck ähnlich. Also gilt für das Verhältnis der Streckenlängen von Ordinaten- und Abszissenabschnitt:
|
![{\displaystyle {\frac {[S]}{v}}=\quad {\frac {1}{v_{\mathrm {max} }}}\cdot [S]+{\frac {K_{\mathrm {m} }}{v_{\mathrm {max} }}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/591e5f9647e45c4bb60ae2a181220b5e354cf774)

![{\displaystyle \textstyle {[S] \over v}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/51466cc83457b16de6b692fbd56fc58f9e95eea1)



Fehler in [S]/v sind eine weit bessere Annäherung der Fehler in v. Aufgrund einer unverfälschten Spreizung der Messpunkte entlang der [S]-Achse wird das Ergebnis durch einzelne Ausreißer prinzipiell weniger verfälscht. Da aber abhängige und unabhängige Variable vermischt werden, ist auch hier eine Datenoptimierung durch lineare Regression nicht sinnvoll.
Hill-Diagramm
BearbeitenDas Hill-Diagramm ist eine Darstellung der Hill-Gleichung, in der (Ordinatenwert) als Funktion von (Abszissenwert) aufgetragen wird. Die zugehörige Umformung der Hill-Gleichung ergibt:

![{\displaystyle \log {[S]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6f478c51e07c55625e5ac74c4fab69f67ca27f63)
- (10)
![{\displaystyle \log \left({\dfrac {\theta }{1-\theta }}\right)=n_{H}\log {[S]}-\log {K_{D}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/7be3a4cc37ed5921dbb42506fc967f3fd16c9a48)
| Umformung |
|
Die Hill-Gleichung wird nach Ersetzen vom durch gemäß Überlegung (9) zu: Übergang zur reziproken Zahl
Brüche stürzen zu einer wählbaren Basis; mit der Rechenregel für den Logarithmus eines Quotienten bzw. den Logarithmus einer Potenz: (10) |
![{\displaystyle \theta ={\frac {[S]^{n_{H}}}{K_{D}+[S]^{n_{H}}}};\quad \mid }](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc9c816d65b360dec56e0116b94c6885f797517b)
![{\displaystyle {\frac {1}{\theta }}={\frac {K_{D}+[S]^{n_{H}}}{[S]^{n_{H}}}}={\frac {K_{D}}{[S]^{n_{H}}}}+1;\quad \mid -1}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6824c602ff3207d2ff55fa939f1873e9f7fd3972)
![{\displaystyle {\frac {1}{\theta }}-1={\frac {1-\theta }{\theta }}={\frac {K_{D}}{[S]^{n_{H}}}};\quad \mid }](https://wikimedia.org/api/rest_v1/media/math/render/svg/e3bf3daae543e21314d3c656afdf91a7bde251a4)
![{\displaystyle {\frac {\theta }{1-\theta }}={\frac {[S]^{n_{H}}}{K_{D}}};\quad \mid \log }](https://wikimedia.org/api/rest_v1/media/math/render/svg/3c085aef3dae8933c5dbdd7372a316c7b2b67d73)
![{\displaystyle \log \left({\frac {\theta }{1-\theta }}\right)=\quad \log \left({\frac {[S]^{n_{H}}}{K_{D}}}\right)=\quad \log \left([S]^{n_{H}}\right)-\log K_{D}=\quad n_{H}\cdot \log[S]-\log K_{D};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/edd61d85274930b84ce8b34a55db7450b2623db3)
Bei Verwendung von hat (10) die Form:
- (10a)
![{\displaystyle \log \left({\dfrac {\theta }{1-\theta }}\right)=n_{H}\log {[S]}-n_{H}\log {K_{A}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/3f14405be673f963c268e4b308130f8979a3888f)
| Umformung |
|
Die Definition der Halbsättigungskontante in Gleichung (3') wird nach Ersetzen vom durch gemäß Überlegung (9) zu:
mit der Rechenregel für den Logarithmus einer Potenz: dies kann in (10) eingesetzt werden. |


Auch in den hier folgenden Gleichungen kann an die Stelle von treten.


Ist bekannt, so lassen sich die Ordinatenwerte unter Verwendung von bestimmen:
- (10b)
![{\displaystyle \log \left({\dfrac {v}{v_{\mathrm {max} }-v}}\right)=n_{H}\log {[S]}-\log {K_{D}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/5da1310da072faf944dc1eae587a3939b04b7efc)
| Umformung |
|
Die Gleichungen (10) und (10b) sind äquivalent, wenn das Argument des Logarithmus der linken Seite für beide Gleichungen übereinstimmt. Einsetzen von Gleichung (4) und Erweitern mit ergibt: |

Die Sättigungsfunktion kann in die Gleichung eingeführt werden:
- (10c)
![{\displaystyle \log \left({\dfrac {r}{n_{H}-r}}\right)=n_{H}\log {[S]}-\log {K_{D}};\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2f2e6ab85751ec801bb76f336a1ce79cde43db1e)
| Umformung |
|
Die Definition der Sättigungsfunktion nach Gleichung (6) wird nach Ersetzen vom durch gemäß Überlegung (9) zu:
die Gleichungen (10) und (10c) sind äquivalent, wenn das Argument des Logarithmus der linken Seite für beide Gleichungen übereinstimmt. Erweitern mit und Einsetzen von (i) ergibt: |


Insoweit die Hill-Gleichung eine Enzymkinetik zutreffend beschreibt, zeigt das Hill-Diagramm eine Gerade , aus der sich

- als deren Steigung und
- als Abszissenabschnitt (= Abszissenwert des Schnitts von mit der Abszisse)

ablesen lässt; hieraus lässt sich nach Delogarithmieren und nach Berechnung von und anschließendem Delogarithmieren auch bestimmen.

| Begründung und Rechenhinweise |
|
A. Diejenigen Varianten der Gleichung (10), die den Summanden enthalten, haben die Form
und sind daher als Funktionsvorschrift einer linearen Funktion mit Steigung deutbar, deren Graph die angegebene Gerade ist. B. Zur Bestimmung des Abszissenabschnitts einer solchen linearen Funktion ist ihr Ordinatenwert null zu setzen: wie im Text angegeben. Bemerkung: Diejenigen Varianten der Gleichung (10), die den Summanden enthalten, sind Geradengleichungen der Form:
Aus mathematischer Sicht könnte zur hier beschriebenen Rechnung eine beliebige Basis verwendet werden, üblich sind jedoch vor allem
jeweils entsprechend zum Delogarithmieren von bzw. . Im angelsächsischen Sprachraum wird nicht nur der allgemeine, sondern auch der natürliche Logarithmus zuweilen mit bezeichnet (was zu Verwechselungen führen kann). |


![{\displaystyle \log {[S]}=(\log[S])_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1010afe960b37792cd28fb7fdf71a2480fa5dc9b)
![{\displaystyle 0=\quad n_{H}\cdot (\log[S])_{0}-\log {K_{D}}=\quad n_{H}\cdot (\log[S])_{0}-n_{H}\cdot \log {K_{A}};\quad \mid -n_{H}\cdot (\log[S])_{0}\quad \mid :(-n_{H})\neq 0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/015cfd028094cce2cff2b7c48dd7086fb9b96ab9)
![{\displaystyle (\log[S])_{0}=\log {K_{A}},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a7b82f613232d72b7ae6917735f86ad78b478c2f)













Im nebenstehenden Hill-Diagrammen[12] ist die Abszissenvariable mit bezeichnet, der Abszissenabschnitt mit , die Ordinatenvariable mit . (Die Längeneinheit ist für beide Achsen unterschiedlich gewählt, sodass die Steigung der roten Gerade nicht ist. Der Schnitt einer Gerade mit der eingezeichneten Ordinate, die nicht durch den Nullpunkt der Abszisse führt, ist nicht der Ordinatenabschnitt der jeweiligen Gerade.)
![{\displaystyle \textstyle \log {[S]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/22c7865d7c15af8d8e7ea6b734e2a00c755a6d01)
![{\displaystyle \textstyle \log {[X]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f4111f9bc84865192d54ee04d0adb7c921f1bade)





Für den gleichen Abszissenabschnitt (und damit den für beide Geraden gleichen Wert von ) ist

- die rote Gerade das Hill-Diagramm eines hochkooperativen Enzyms (Schnitt mit der Ordinate bei Geradensteigung );
- die grüne Gerade dasjenige eines kaum oder nicht kooperativen Enzyms (Schnitt mit der Ordinate bei Geradensteigung ).




Wenn die berechneten Wertepaare nicht auf einer Gerade liegen, kann diese außer durch zufällige auch durch systematische Fehler bedingt sein, denn die Hill-Gleichung setzt voraus, dass der Hill-Koeffizient für alle Konzentrationen gleich ist. Eine Abweichung hiervon fand G.S. Adair, der ebenfalls die Sauerstoffbindung von Hämoglobin untersuchte.[13]
Zusammenstellung von Linearisierungen einer Hyperbel
Bearbeiten

Inhibitoren
BearbeitenHauptartikel: Enzymhemmung
Viele Therapeutika und Gifte sind Hemmstoffe (Inhibitoren) von Enzymen. Aus diesem Grunde ist der Aufklärung des Wirkungsmechanismus immer eine besondere Bedeutung zugekommen. Die Nomenklatur der Hemmtypen wurde von William Wallace Cleland (* 1930) 1963 auf eine systematische Grundlage gestellt, leider werden in vielen Lehrbüchern immer noch Begriffe abweichend verwendet.
Hier sollte allerdings beachtet werden, dass sich klassische Analysen auf reversibel bindende Stoffe beschränken. Irreversible Bindung einer Substanz an ein Enzym führt zur Inaktivierung, nicht zur Hemmung.
Abgeleitet aus der Michaelis-Menten-Gleichung stellt sich die allgemeine Inhibitionsgleichung wie folgt dar:
![{\displaystyle v=V_{\text{max}}\cdot [\mathrm {S} ]/(K_{m}+[{\textrm {S}}])}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b5cb620c22bf1e468edac97061acff730b7a54cb)
![{\displaystyle v={\frac {V_{\text{max}}\cdot [\mathrm {S} ]}{K_{m}(1+{\frac {[\mathrm {I} ]}{K_{i}}})+[\mathrm {S} ](1+{\frac {[\mathrm {I} ]}{K_{ii}}})}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a76f5febb27ee818d773e5e5bf6584a697ed86c5)
Danach kann das Verhältnis des -Wertes (Dissoziationskonstante des Komplexes EI) und des -Wertes (Dissoziationskonstante des Komplexes EIS) zur Ableitung des Inhibitionstyps dienen:


Kompetitiv
Bearbeiten
Inhibitor und Substrat schließen sich gegenseitig von der Bindung an das Enzym aus. Dies bedeutet jedoch nicht notwendigerweise, dass der Inhibitor an der gleichen Bindungsstelle bindet wie das Substrat. Auch wenn die Bindung von Substrat bzw. Inhibitor zu Konformationsänderung im Enzym führen, welche die Bindungsstelle für den jeweils anderen blockieren, ist die Hemmung kompetitiv. Wenn Substrat und Inhibitor allerdings die gleiche Bindungsstelle haben, dann ist der Hemmtyp notwendig kompetitiv.
Bei der Kompetitiven Hemmung kann der Inhibitor durch Substrat aus dem Enzym verdrängt werden, ändert sich also nicht. Allerdings wird für jede gewünschte Geschwindigkeit eine höhere benötigt, die scheinbare wird also mit steigender höher. Im Lineweaver-Burk-Diagramm führt dies bei unterschiedlichen beziehungsweise zu einer Schar von Geraden, die einen gemeinsamen Schnittpunkt auf der y-Achse bei ( ) haben.

![{\displaystyle [\mathrm {S} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e2013f31cdd12d83f4678d2818d446c0c8d3c6d3)
![{\displaystyle [\mathrm {I} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc6388955e17bc40b464954c2143e48767a4d689)


Unkompetitiv
BearbeitenDer Inhibitor bindet nicht an das freie Enzym, sondern an den ES-Komplex. Höhere Konzentrationen des Substrates können daher den Hemmstoff nicht vom Enzym verdrängen, sondern führen zu vermehrter Bindung. Umgekehrt vermindert Bindung des Hemmstoffes die Konzentration von ES, nach dem Prinzip von Le Chatelier muss sich also zusätzliches ES aus E und S bilden: Die scheinbare vermindert sich, die Affinität des Enzymes für das Substrat steigt mit steigender . Gleichzeitig nimmt natürlich ab. Im Lineweaver-Burk-Diagramm finden wir eine Schar paralleler Geraden.
Nicht-kompetitiv
BearbeitenDer Inhibitor kann sowohl an E als auch an ES binden. Im einfachsten Fall ist dabei , d. h., dass die Substratbindung die Affinität des Enzymes für den Inhibitor nicht verändert, etwa durch Konformationsänderung. Dann folgt natürlich auch, dass die Bindung des Inhibitors die Affinität des Enzymes für das Substrat nicht ändert und . Wegen des Zusammenhangs zwischen und ändert die Bindung von Inhibitor also auch nicht .



Es lässt sich nun zeigen (durch Substitution und Eliminierung aus den Definitionen von und ), dass . Wenn also , dann folgt und die scheinbare steigt mit . Falls andererseits , dann folgt und die scheinbare sinkt mit steigendem .







Die nicht-kompetitive Hemmung führt im Lineweaver-Burk-Diagramm zu einer Schar von Geraden mit gemeinsamen Schnittpunkt links von der y-Achse, der Schnittpunkt liegt auf der x-Achse wenn , er liegt über der x-Achse falls und unter der x-Achse falls .
Gemischt-kompetitive Hemmung
BearbeitenDer Mechanismus dieses Hemmtyps (der in der Praxis von geringer Bedeutung ist) ähnelt der nicht-kompetitiven Hemmung, allerdings hat der EIS-Komplex noch eine katalytische Aktivität. Auch das Lineweaver-Burk-Diagramm sieht aus wie bei der nicht-kompetitiven Hemmung (mit allen 3 Möglichkeiten). Im sog. Sekundärdiagramm (Steigung bzw. y-Schnittpunkt im Lineweaver-Burk-Diagram als Funktion von ) sieht man aber im Falle der nicht-kompetitiven Hemmung Geraden, im Falle der gemischt-kompetitiven jedoch Kurven.
Siehe auch
BearbeitenLiteratur
Bearbeiten- H. Bisswanger: Enzymkinetik – Theorie und Methoden. 3. Auflage. Wiley-VCH, Weinheim 2000, ISBN 978-3-527-30096-9.
- E. Buxbaum: Fundamentals of protein structure and function. Springer, New York 2007, ISBN 978-0-387-26352-6.
- G. E. Briggs, J. B. Haldane: A Note on the Kinetics of Enzyme Action. In: Biochemical Journal. Band 19, Nr. 2, 1925, S. 338–229; PMID 16743508.
- W. W. Cleland: The kinetics of enzyme-catalyzed reactions with two or more substrates or products. In: Biochimica et Biophysica Acta. Band 67, 1963, S. 104–137, 173–187, 188–196.
- R. Eisenthal, A. Cornish-Bowden: The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. In: Biochemical Journal. Band 139, Nr. 3, 1974, S. 715–720; PMID 4854723.
- J. B. S. Haldane: Graphical methods in enzyme chemistry. In: Nature. Band 179, 1957, S. 832.
- V. Henri: Theorie generale de l’action de quelques diastases. In: Comptes rendues l’Academie des sciences. Band 135, 1902, S. 916–919.
- A. V. Hill: The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. In: The Journal of Physiology Band 40, Supplement, 1910, S. iv–vii.
- L. Michaelis, M. L. Menten: Die Kinetik der Invertin-Wirkung. In: Biochemische Zeitschrift. Band 49, 1913, S. 333–369.
- I. H. Segel: Enzyme Kinetics. Wiley, New York 1975 (Nachdruck 1993).
- S. P. L. Sørensen: Enzymstudien II. Über die Messung und Bedeutung der Wasserstoffionenkonzentration bei enzymatischen Prozessen. In: Biochemische Zeitschrift. Band 21, 1909, S. 131–304.
Weblinks
Bearbeiten- Peter Birch: An Introduction to Enzyme Kinetics. ( vom 28. April 2009 im Internet Archive) Department of Biological Sciences, University of Paisley.
- Private Internetseite zur Enzymkinetik
Einzelnachweise
Bearbeiten- ↑ Leonor Michaelis, Maud Leonora Menten: Die Kinetik der Invertinwirkung. In: Biochemische Zeitschrift. Band 49, 1913, ISSN 0366-0753, S. 333–369, urn:nbn:de:hebis:30-1090119.
- ↑ Die Darstellung orientiert sich in freier Formulierung der hier genannten Quelle.
- ↑ A.V. Hill, "The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves", J Physiol 1910, 40, S. iv–vii
- ↑ vgl. z. B. Jachen Denoth: „Theoretische Betrachtungen zur maximalen Leistung im Ausdauersport bei einem durch die maximale Sauerstoffaufnahme begrenzten Energieverbrauch“ in: Schweizer Zeitschrift für Sportmedizin und Sporttraumatologie 2008, 65 (2), S. 77–81
- ↑ z. B. zur Beschreibung einer Michaelis-Menten-Gleichung in: Hartmut Bossel: "Modellbildung und Simulation", Springer-Verlag, 13. März 2013, S. 271
- ↑ Hans Bisswanger: „Enzyme: Struktur, Kinetik und Anwendungen“, John Wiley & Sons, 29. Juni 2015, Box 11.2
- ↑ Berg/Stryer/Tymoczko: Biochemie, S. 214, Springer-Verlag, 27. Februar 2015.
- ↑ Schemazeichnung für die aufgeführten drei Möglichkeiten in https://images.slideplayer.org/1/649096/slides/slide_39.jpg
- ↑ Das Diagramm ist der englischsprachigen Wikipedia (Seite: Hill equation (biochemistry)) entnommen, der Kommentar unabhängig formuliert.
- ↑ H. Lineweaver, D. Burk: The Determination of Enzyme Dissociation Constants. In: Journal of the American Chemical Society, 56, 1934, S. 658–666. doi:10.1021/ja01318a036.
- ↑ Hanes, CS. (1932). Studies on plant amylases: The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. In: Biochemical Journal 26: 1406–1421; PMID 16744959; PMC 1261052 (freier Volltext).
- ↑ Das Diagramm ist der englischsprachigen Wikipedia (Seite: Hill equation (biochemistry)) entnommen, der Kommentar unabhängig formuliert.
- ↑ 'The hemoglobin system. IV. The oxygen dissociation curve of hemoglobin' in: J Biol Chem 63, 1925, S. 529–545
{kind=link}